
Вопросы развития постинфарктного ремоделирования сердца и важности нейрогуморальной активации в этих процессах. Роль альдостерона. Доказательства отрицательного влияния повышенной концентрации альдостерона в крови на течение сердечно-сосудистых заболеваний и развитие сердечной недостаточности после перенесенного инфаркта миокарда. Данные об эффективности и безопасности применения препаратов из группы антагонистов альдостерона, полученные в ходе нескольких рандомизированных клинических исследований. Клиническое применение селективного блокатора рецепторов альдостерона эплеренона.
М.Г.Бубнова ФГБУ Государственный научно-исследовательский центр профилактической медицины Минздравсоцразвития РФ, Москва
Резюме.
В обзоре литературы рассматриваются вопросы развития постинфарктного ремоделирования сердца и важности нейрогуморальной активации в этих процессах. Особое внимание уделяется роли альдостерона. Представлены доказательства отрицательного влияния повышенной концентрации альдостерона в крови на течение сердечно-сосудистых заболеваний и развитие сердечной недостаточности после перенесенного инфаркта миокарда. Приведены данные об эффективности и безопасности применения препаратов из группы антагонистов альдостерона, полученные в ходе нескольких рандомизированных клинических исследований. Особое место уделяется аспектам клинического применения селективного блокатора рецепторов альдостерона эплеренона, а также данным, обосновывающим его использование в клинической практике у больных после острого инфаркта миокарда с левожелудочковой недостаточностью.
Ключевые слова: инфаркт миокарда, ремоделирование сердца, антагонисты альдостерона, эплеренон.
В 2010 г. в России острый коронарный синдром (ОКС) перенесли 594 583 человека [1]. Заболеваемость первичным острым инфарктом миокарда (ОИМ) в 2010 г. составила на 100 тыс. населения 134,0 случая (против 140,2 в 2008 г.), а частота повторного инфаркта миокарда (ИМ) – на 100 тыс. населения – 25,6 случая (против 21,2 в 2008 г.). Как ни парадоксально, но улучшение лечения ОКС ведет к повышению не только выживаемости пациентов, но и к их «доживанию» до серьезных сердечно-сосудистых осложнений. Эта ситуация описана как «парадокс в улучшении лечения сердечно-сосудистых заболеваний» (ССЗ), или «ironic failure of success».
К моменту выписки из клиники пациента, перенесшего ОИМ, патологические процессы в миокарде не заканчиваются. Остается высоким риск развития внезапной сердечной смерти (ВСС) – ее частота составляет более 30% случаев, риск мозгового инсульта возрастает в 3–4 раза, риск дилатации левого желудочка (ЛЖ) – в 50–65% случаев с последующим развитием клинической сердечной недостаточности (СН), увеличивающей риск смерти в 3 раза [2, 3].
ОИМ – стартовая точка для развития СН, повышающей риск смерти пациента в более ранние сроки. По данным исследований, от 20,4 до 33% пациентов с ОИМ имели клиническую СН уже при поступлении в стационар (класс II и выше по Killip), а у 38% пациентов она развилась в течение первых 5 дней [4–6]. Эпизоды СН в течение 30 дней после ИМ возникали у 59% пациентов [7].
В Глобальном реестре ОКС GRACE (Global Registry of Acute Coronary Events, 11 543 пациентов из 14 стран) СН при поступлении в стационар выявлялась у 15,6% пациентов с ИМ с подъемом сегмента ST (ИМпST), у 15,7% пациентов с ИМ без подъема сегмента ST и у 8,2% пациентов с нестабильной стенокардией [8]. У пациентов с ИМ, осложненным СН, госпитальная летальность в 4 раза выше относительно пациентов с ИМ без признаков СН, а смертность в последующие 6 мес – выше в 3 раза. Примерно каждый восьмой пациент с низкой фракцией выброса (ФВ) ЛЖ (<40%) умирал в пределах 16 мес после ОИМ [9]. Таким образом, пациент после ОИМ может умереть внезапно либо вследствие ремоделирования сердца, спровоцированного развитием «оглушенного» миокарда (недостаточно кровоснабжаемого и не сокращающего, хотя и живого), либо от острой СН. В связи с этим, важное значение приобретает грамотное ведение пациента после выписки из стационара на амбулаторно-поликлиническом этапе.
Процессы постинфарктного ремоделирования сердца
В конце 1970-х гг. N.Sharp ввел термин «ремоделирование сердца», подразумевающий структурногеометрические изменения ЛЖ после ОИМ и включающий в себя гипертрофию миокарда, дилатацию полостей и нарушение систолической/диастолической функции [10]. В структурногеометрическую перестройку при ИМ активно вовлекаются, как инфарктная зона, так и неинфарцированные участки миокарда. Геометрический тип ремоделирования сердца в ответ на развитие ИМ сочетает патогенетические механизмы, обусловленные перегрузкой давлением и объемом, а также непосредственным поражением миокарда. Цель постинфарктного ремоделирования сердца (ПРС) – его адаптация к новым условиям функционирования в ранний и последующий периоды после ОИМ. Процесс ремоделирования сердца начинается с первых часов ИМ и продолжается в течение многих месяцев.
Ранняя фаза ПРС (первые 72 ч ОИМ) включает непропорциональное растяжение клеток и истончение стенки ЛЖ в зоне ИМ вследствие соскальзывания (slip-page) мышечных волокон друг относительно друга за счет ослабления связей между кардиомиоцитами – КМЦ (в результате деградации коллагенового матрикса) и снижения плотности их расположения в инфарктной зоне. Некротизированный участок миокарда не в силах противостоять внутрижелудочковому давлению, он выпячивается [11]. Данный процесс, описанный в 1978 г. G.Hutchins и B.Bulkley, был назван «экспансией инфаркта», т.е. острым увеличением и истончением инфарктной зоны, не связанными с дополнительным ишемическим некрозом миокарда. Чаще всего экспансия развивается после трансмурального ИМ (более вероятна для экспансии передневерхушечная область, поскольку она наиболее изогнута), и ее выраженность зависит от особенностей внутрисердечной гемодинамики. Пациенты с экспансией инфаркта угрожаемы по риску серьезных осложнений, таких как СН, аневризма и разрыв миокарда.
Некроз клеток в результате ИМ инициирует воспалительную реакцию с лейкоцитарной инфильтрацией и высвобождением протеолитических ферментов, в частности матриксных металлопротеиназ (ММП), расщепляющих молекулы коллагена [12]. Распад коллагена преобладает над его образованием вплоть до 14-го дня с момента ИМ, и на протяжении этого периода концентрация ММП типа 1 остается повышенной. Далее превалирует активность ингибитора ММП типа 1, и процесс смещается в сторону отложения коллагена и формирования рубца в зоне ИМ.
После повреждения и гибели части КМЦ в интактном миокарде активизируются процессы, направленные на поддержание сердечного выброса (СВ) по закону Франка–Старлинга и системной гемодинамики. После ИМ размер ЛЖ увеличивается, и меняется радиус кривизны стенок, т.е. происходит сферификация ЛЖ с развитием адаптивной тоногенной дилатации. Для выброса увеличенного остаточного объема крови из дилатированного ЛЖ ему необходимо развивать значительно большее напряжение. Это достигается гипертрофией неповрежденной инфарктом сердечной мышцы, обусловленной перегруппировкой миофибрилл – увеличением числа последовательно расположенных саркомеров (феномен «соскальзывания» мышечных волокон) или удлинением (склеиванием по типу «конец в конец») КМЦ. Наблюдается нарастание мышечной массы ЛЖ без утолщения его стенок, т.е. формируется эксцентрическая гипертрофия миокарда. Между давлением в ЛЖ и его объемом устанавливаются новые соотношения, позволяющие определенное времени сохранять ударный объем сердца, несмотря на уменьшение ФВ ЛЖ.
Изначально ПРС представляет собой компенсаторный процесс, направленный на поддержание сократительной функции ЛЖ за счет расширения камер сердца и гипертрофии миокарда. Эффективность компенсации зависит от размера и локализации ИМ, объема жизнеспособного миокарда, выраженности гипертрофии КМЦ и интерстициальных фиброзных изменений, а также от состояния коронарного кровотока уцелевшего миокарда. При неадекватном кровоснабжении миокарда дилатация полости ЛЖ выражена в большей степени. Клинические симптомы СН после ИМ развиваются практически у всех пациентов с поражением более 20–30% площади ЛЖ. Прирост массы миокарда относительно прогрессирующей постинфарктной дилатации происходит не пропорционально. Возможности ЛЖ преодолевать возросшие нагрузки постепенно исчерпываются, нарушается хрупкий баланс и развивается миогенная дилатация с гемодинамически невыгодной сферической формой ЛЖ. Это ускоряет прогрессирование болезни, ухудшает шансы пациента на выживание, и тогда от гибели пациента могут отделять уже лишь месяцы.
ПРС – процесс сложный, тесно связанный с активацией симпатико-адреналовой системы (САС) и ренин-ангиотензин-альдостероновой системы (РААС), а также с нарастанием дисфункции эндотелия (ДЭ) сосудов. Активация САС и РААС важна для адаптации работы сердца в новых условиях и направлена на поддержание СВ и артериального давления (АД), стимуляцию ранней гипертрофии КМЦ и замещение зоны ИМ рубцовой тканью. В дальнейшем эти компенсаторные механизмы приобретают патологический характер и негативно влияют на ПРС и развитие СН.
Неблагоприятное последствие активации САС – это повышение напряжения стенки ЛЖ и связанное с этим усугубление экспансии инфаркта, повышение потребности миокарда в кислороде и проаритмогенный эффект катехоламинов. Эффекты активации РААС, повышающие постнагрузку, внутрижелудочковое давление и тем самым неблагоприятно влияющие на процессы ПРС, – это вазоконстрикция и задержка жидкости. Ангиотензин II (АТ II), образующийся в миокарде под влиянием тканевой РААС, повышает проницаемость эндотелия коронарных артерий, облегчая диффузию ростовых факторов к месту их действия, регулирует процессы апоптоза (запрограммированной гибели клеток), активирует митогены и факторы роста, участвующие в процессах ПРС, стимулирует продукцию цитокинов и других нейрогормонов (альдостерона, вазопрессина, эндотелина).
Известно, что альдостерон стимулирует синтез коллагена фиб-робластами и играет роль в гибели КМЦ через влияние на внутриклеточный баланс электролитов. В свою очередь активация фибробластов в ответ на ишемическое повреждение приводит к фиброзу миокарда в зоне инфаркта и здоровых участков, увеличению жесткости стенок ЛЖ, диастолической дисфункции, нарушению передачи электрических импульсов и апоптозу. Все эти процессы ускоряют прогрессирование СН, увеличивают электрическую гетерогенность миокарда, лежащую в основе механизмов re-en-try и снижения порога возникновения жизненно опасных желудочковых нарушений ритма сердца.
Апоптоз, как и некроз, ведет к дальнейшему нарушению структуры (быстрой потере сократительных элементов) и функции ЛЖ. Известными триггерами апоптоза являются цитокины, оксидативный стресс и повреждение митохондрий. Так, в сердце больных, умерших от ОИМ, в течение первых 10 дней апоптоз выявляется в 12% миоцитов, расположенных на границе инфаркта, и в 1% клеток в зоне, удаленной от ИМ.
Для эффективного воздействия на процессы ПРС и реального улучшения прогноза пациентов терапию необходимо начинать как можно раньше. Доказано, что раннее назначение препаратов, блокирующих активность САС – b-адреноблокаторов (БАБ) и РААС – ингибиторов ангиотензинпревращающего фермента (ИАПФ), блокаторов рецепторов АТ II (БРА), а также антагонистов альдостерона (АА), реально повышает выживаемость больных после ОИМ.
Альдостерон и сердечно-сосудистые механизмы его влияния
Альдостерон – минералокортикоидный гормон, секретируемый корой надпочечников (преимущественно в zona glomerulosa коры надпочечников)[13]. Секреция альдостерона увеличивается в ответ на активацию РААС, повышение синтеза АТ II и уровня калия плазмы крови (рис. 1). Воздействуя на минералокортикоидные рецепторы (МР) – ядерные рецепторы в эпителиальных (в том числе в дистальных почечных канальцах) и неэпителиальных (в том числе в сердце, сосудах, головном мозге) тканях, альдостерон увеличивает уровень АД, реабсорбцию натрия и задержку жидкости в дистальном отделе нефрона, одновременно снижая уровень калия [14].

Первое влияние альдостерона на миокард было описано в 1988 г. группой исследователей под руководством K.Weber, работавших в Чикаго. Показано, что уже через 3–6 мин от начала инфузии альдостерона повышается периферическое сосудистое сопротивление, изменяется барорефлекторная чувствительность и увеличивается СВ [15, 16]. Такие «быстрые эффекты» альдостерона связывают с его воздействием на мембранные (неядерные) МР, активацией «быстрых» кальциевых каналов и повышением экспрессии протеин-киназы С [17]. «Быстрые эффекты» альдостерона не блокируются классическим АА спиронолактоном, но они чувствительны к новому селективному АА эплеренону [18].
Высказывается мнение, что именно локально синтезированный и фиксированный к мембранным рецепторам сердца альдостерон играет важную роль в процессах ПРС [19, 20]. В ряде работ продемонстрирована прямая корреляция уровня альдостерона с массой миокарда. [21, 22]. В эксперименте на животных показано, что увеличение концентрации альдостерона в крови приводит к тяжелому повреждению сердца, включая развитие периваскулярного воспаления, микроинфарктов и фиброза [23, 24]. С одной стороны, альдостерон стимулирует биосинтез и накопление коллагена 1го и 3-го типов фибробластами в сердце (развивается репаративный фиброз), с другой стороны, инициирует воспалительные изменения в сосудистой стенке (альдостерон-индуцированная васкулопатия) в виде моноцитарно-макрофагальной инфильтрации, увеличения активности медиаторов воспаления (циклооксигеназы-2, макрофагального белкового хемоатрактанта типа 1 и остеопонтина) [25–28].
Гиперальдостеронизм влечет за собой снижение числа сократительных элементов в единице объема миокарда с развитием гипоксии КМЦ и нарушением их синхронной работы. Кроме того, при гиперпродукции альдостерона увеличивается плотность АТ-рецепторов типа 1 и рецепторов к эндотелину, индуцируется экспрессия АПФ в КМЦ, что обеспечивает дополнительную стимуляцию биосинтеза коллагена [29, 30]. Индуцируя активность ММП через разные механизмы, альдостерон участвует в ПРС [31]. Один из механизмов негативного влияния альдостерона на структуру миокарда – это его способность активировать апоптоз КМЦ [32]. Альдостерон может влиять на развитие ССЗ через формирование ДЭ [33]. Развитие альдостерон-индуцированной ДЭ связывают со снижением биодоступности оксида азота (NO) и активности NO-синтазы [34]. На клеточном уровне альдостерон изменяет сигналы от ядерного фактора NF-kB, индуцирует оксидативный стресс и доступность активных форм кислорода в сосудистую стенку, что имеет значение в развитии ДЭ [35, 36]. Повышение синтеза альдостерона усиливает активность ингибитора активатора плазминогена типа 1 (ИТАП-1), что приводит к коагулопатии, микротромбозу и последующему фиброзу почечных артериол [37, 38].
Альдостерон, риск ИМ и его осложнений
У пациентов с ОИМ уже в первые часы выявляются высокие уровни альдостерона в моче и плазме, а также ренина (максимально к 3-му дню). В пораженном участке миокарда и периинфарктной зоне возрастают экспрессия АПФ и содержание АТ II. По данным Н.В.Сытник и соавт., у пациентов с первичным ИМпST при выписке из стационара высокие уровни альдо-стерона встречались чаще (58%), чем повышение ренина (39%) и АТ II (9%) [39].
Определено, что у пациентов после ИМ с высокой (≥141 пг/мл) концентрацией альдостерона в крови риск смерти в течение 5 лет наблюдения был в 2 раза выше, чем при низкой (<83,2 пг/мл) концентрации [40]. В другом исследовании у больных с ИМпST установлена прямая связь уровня альдостерона в крови с госпитальной летальностью, нарастанием симптомов СН, риском фибрилляции желудочков и ВСС [41]. После ОИМ активация синтеза альдостерона в миокарде увеличивается за счет повышения активности альдостерон-синтазы mRNA (в 2 раза), через стимуляцию АТрецепторов типа 1 (в 3,7 раза) и вследствие повышения кардиального уровня АТ II (в 1,9 раза). Влияние альдостерона на риск осложнений при ОИМ связывают как с его прямыми (геномными) эффектами воздействия на водно-электролитный баланс, так и с влиянием на ПРС, функцию эндотелия, процессы воспаления и антиоксидантную систему.
Субстадия исследования VALIANT (Valsartan in Acute Myocardial Infarction Trial) показала, что у 14 609 пациентов после ОИМ в первые 30 дней после выписки из стационара ВСС развилась у 83% несмотря на терапию БАБ, ИАПФ и/или БРА [42]. В исследовании CONSENSUS (Cooperative North Scandinavian Enalapril Survival Study) пациенты с высоким уровнем альдостерона в крови в течение 6 мес имели достоверно выше смертность, чем с низким уровнем (55% против 32%, р<0,001), т.е. исходные уровни альдостерона могут рассматриваться как предикторы выживаемости пациентов после ОИМ [43].
Стойкое повышение концентрации альдостерона в крови наблюдается при формировании СН, в период ее клинического проявления и является предиктором неблагоприятных исходов у пациентов с хронической СН (ХСН) вне зависимости от ее функционального класса (ФК) по NYHA [44–46].
Эффективное подавление активности АПФ посредством ИАПФ у больных с СН существенно не снижало концентрацию альдостерона в крови (у 40% пациентов она была выше 144 пг/мл) вследствие развития феномена «ускользания» блокады синтеза альдостерона – он развивался у 20–50% пациентов [47–49]. По данным B.Pitt, увеличения уровня альдостерона на фоне приема ИАПФ можно ожидать уже через 3 мес [50]. По-видимому, это обусловлено неполной блокадой АПФ (имеются разные пути его синтеза) и уменьшением катаболизма альдостерона в печени, особенно у больных с ХСН.
Слабое подавление активности альдостерона – одна из возможных причин недостаточной эффективности стандартной терапии в лечении ССЗ. Устранение негативных эффектов альдостерона в сердце сопровождается блокадой процессов ПРС ЛЖ, снижением натрийуретических пептидов – известных маркеров прогноза при СН [51].
Фармакологические особенности селективного АА эплеренона
Первый высокоселективный конкурентный АА эп-леренон имеет ряд явных преимуществ перед классическим неселективным АА спиронолактоном. Эпле-ренон обладает меньшим сродством к рецепторам прогестерона (менее 1%) и андрогеновым рецепторам (менее 0,1%), чем спиронолактон. Этим объясняются лучшая его переносимость и меньшая частота развития эндокринологических осложнений (в виде гинекомастии и импотенции у мужчин или дисменореи у женщин), свойственных спиронолак-тону [52]. Эплеренон разрушается печенью (почками выводится менее 5% препарата), не имеет активных метаболитов, период полувыведения составляет 4– 6 ч. После приема препарата концентрация в плазме крови пиковая – через 1,5 ч и устойчивая – через 2 дня. Доза эплеренона in vivo, требуемая для адекватной альдостероновой блокады, составляет 50–75% от дозы спиронолактона [53].
Эффективность эплеренона при артериальной гипертонии (АГ) была изучена в нескольких многоцентровых исследованиях с включением более 3 тыс. больных [54].
Кардиопротективная эффективность эплеренона
Первым основанием для включения класса АА в рекомендации по лечению ХСН III–IV ФК в качестве дополнительного средства к основной терапии послужили результаты многоцентрового плацебоконтролируемого исследования RALES (Randomized Aldac-tone Evaluation Study) с применением низких доз (до 50 мг/сут) спиронолактона [55, 56].
Значимость эплеренона для улучшения прогноза жизни пациентов с признаками СН и/или сниженной ФВ ЛЖ после перенесенного ИМ установлена в крупном рандомизированном двойном слепом плацебо-контролируемом исследовании EPHESUS (Eplerenone Post-Acute Myocardial Infarction Heart Failure Efficacy and Survival Study) [57]. В исследование включали (n=6632) пациентов (средний возраст 64 года) с ОИМ (3–14 сут, в среднем на 7,3 сут), осложненным систолической дисфункцией ЛЖ (ФВ ЛЖ ≤40%, средняя ФВ 33%) и/или клиническими признаками СН (у 90% пациентов).
Не включали пациентов, принимающих калийсберегающие диуретики, с плазменной концентрацией креатинина более 2,5 мг/дл (220 мкмоль/л) и калия более 5,0 ммоль/л. В обеих группах 45% больных получили коронарную реперфузионную терапию (тромболизис и чрескожное коронарное вмешательство) и оптимальную медикаментозную терапию (87% – ИАПФ/БРА, 75% – БАБ, 60% – диуретики, 88% – ацетилсалициловая кислота, 47% – статины). Начальная доза эплеренона – 25 мг/сут в течение 4 нед с последующим увеличением до 50 мг/сут (при содержании калия менее 5,0 ммоль/л). Средняя доза эплеренона в исследовании – 42,6 мг/сут, а период наблюдения – 16 мес. Добавление эплеренона (n=3319) к стандартной терапии против плацебо (только стандартная терапия, n=3313) сопровождалось снижением риска смерти от любой причины (на 15%, р=0,008) и риска смертельного исхода/госпитализации по сердечно-сосудистой причине (на 13%, р=0,002). Наиболее значимое снижение смерти от любой причины наблюдалось у пациентов с АГ, высоким пульсовым давлением, низкой концентрацией креатинина и при одновременном приеме ИАПФ и БАБ (рис. 2).
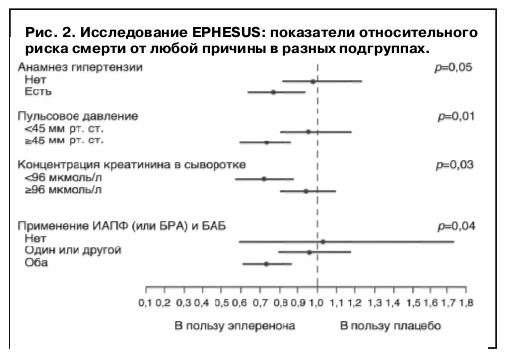
В EPHESUS выявлялось снижение риска ВСС (на 21%, р=0,03), числа первичных (на 15%, р=0,03) и повторных (на 23%, р=0,002) госпитализаций вследствие СН, а также сердечно-сосудистой смертности – ССС (на 17%, р=0,005). В группе эплеренона против группы плацебо снижался (на 8%, р=0,02) суммарный показатель «общая смертность + частота любых гос-питализаций».
Поскольку у пациентов после ИМ, осложненного СН, риск госпитальной смерти в 3–4 раза выше, чем у пациентов без СН, в исследовании EPHESUS анализировалась эффективность эплеренона в снижении ранней смертности – через 30 дней после рандомизации [57]. Добавление селективного АА к стандартной терапии против плацебо к 30-му дню приводило к снижению общей смертности (на 31%, р=0,004) с развитием эффекта уже через 10 дней после рандомизации и риска ССС (на 32%, р=0,003). Особый интерес представлял факт раннего снижения риска ВСС (37% р=0,051). Установлено, что низкая (25 мг/сут) доза эплеренона также обеспечивала заметное снижение смертности и заболеваемости в ранние сроки после ИМ.
Анализ эффективности эплеренона в EPHESUS у пациентов (n=2106) тяжелой подгруппы – с ОИМ и очень низкой исходной ФВ ЛЖ (≤30%) показал существенное снижение общей смертности (на 21%, p=0,012), ССС/частоты госпитализаций (на 21%, p=0,001) и смертности от СН/частоты госпитализа-ций по причине СН (на 25%, p=0,005) [58]. Наиболее впечатляющим у больных с тяжелой левожелудочко-вой дисфункцией было снижение риска ВСС на 58% (р=0,008) через 30 дней от начала терапии эплерено-ном и на 33% (p=0,01) к концу наблюдения.
Госпитализация по причине усугубления течения СН – одно из основных нежелательных явлений для пациентов с СН. Ретроспективный анализ исследования EPHESUS показал меньшее (на 3,6) число дней пребывания в стационаре из-за декомпенсации СН на фоне приема эплеренона, чем на стандартной терапии (13,3 дня против 16,9 дня, р=0,019) [59].
Вероятным объяснением выраженных кардио-протективных эффектов эплеренона у больных ИМ с дисфункцией ЛЖ и признаками СН может быть замедление раннего электрического и структурного ремоделирования ЛЖ [60]. По данным M.Hayashi и соавт., у пациентов с ОИМ, подвергнутых чрескож-ному коронарному вмешательству, АА эффективно предотвращали ПРС и подавляли синтез коллагена в миокарде при назначении в первые 24 ч от развития сосудистого инцидента [61]. Механизмы, посредством которых эплеренон обеспечивает защиту миокарда у пациентов с ОИМ, представлены в табл. 1 [62–64]. R.Rocha и соавт. в эксперименте показали способность эплеренона подавлять миокардиальное и сосудистое поражение, инициированное инфузией альдостерона [65]. Способность АА уменьшать количество желудочковых экстрасистол и даже эпизодов неустойчивой желудочковой тахикардии продемонстрирована в ряде исследований и объясняется, помимо прочего, нормализацией электрического дисбаланса (увеличением концентрации магния и калия) [66]. Улучшение параметров вариабельности сердечного ритма также дополнительно обосновывает возможность АА улучшать прогноз при СН.
Эплеренон не влияет на процессы формирования постинфарктного рубца, напротив, предотвращая реактивный фиброз в участках, отдаленных от зоны ИМ, может препятствовать процессу ПРС и улучшать прогноз жизни пациентов [67].
В плацебо-контролируемом клиническом испытании EMPHASIS-HF (Eplerenone in Mild Patients Hospi-talization and Survival Study in Heart Failure) высокая эффективность эплеренона была продемонстрирована у пациентов с умеренной ХСН II ФК, получавших оптимальную сопутствующую терапию ИАПФ/БРА и БАБ [68].
Присоединение к лечению селективного эплеренона в средней дозе 39,1±13,8 мг/сут (25–50 мг) сопровождалось значительным улучшением клинического течения заболевания и прогноза больных ХСН: снижением комбинированной первичной конечной точки ССС + частоты госпитализаций в связи с декомпенсацией ХСН (на 27%, р<0,001), общей смертности (на 24%, р=0,008), госпитализации по любой причине (на 23%, р<0,001) и в связи с утяжелением СН (на 42%, p<0,001). Таким образом, становится очевидной целесообразность назначения эплеренона пациентам с ХСН при разном ФК (от II до IV).
Безопасность и переносимость эплеренона
Частота нежелательных явлений при применении эплеренона, как правило, не превышает таковую при приеме плацебо. Гинекомастия у мужчин и дисменорея у женщин на фоне эплеренона встречаются реже (<1% случаев), чем на спиронолактоне (10%). Некоторое повышение уровня АД, наблюдаемое в исследовании EPHESUS, было менее выраженным на эплереноне (на 2,4±0,4/1,7±0,2 мм рт. ст.), чем на плацебо (4,0±0,4/2,9±0,2 мм рт. ст.).
В силу своего механизма действия АА, прежде всего в повышенных дозах, увеличивают концентрацию калия в плазме крови. В этом отношении применение эплеренона достаточно безопасно (данные EPHESUS на дозе 50 мг/сут): через год гиперкалиемия (>6,0 ммоль/л) развивалась у 5,5% больных на эплереноне против 3,9% больных на плацебо (р=0,002) и чаще встречалась при низком клиренсе креатинина (<50 мл/мин), начальной гиперкалиемии (>5,5 ммоль/л). Главный вывод post hoc-анализа результатов EPHESUS: гиперкалиемия, развивающаяся в ответ на терапию эплереноном, предсказуема, управляема и нефатальна.
На фоне лечения АА появлению гиперкалиемии способствуют дисфункция почек, сахарный диабет с микроальбуминурией, прием нестероидных противовоспалительных препаратов, высокие дозы ИАПФ/БРА. Определять концентрацию калия в плазме крови целесообразно на 1-й неделе (на 3-й и 7-й дни) от начала терапии, через 1 нед от изменения дозы, через месяц и далее 1 раз в 3 мес при постоянном приеме. Подбор дозы эплеренона в зависимости от сывороточной концентрации калия представлен в табл. 2 (по данным исследования EPHESUS) [69].
При назначении больным ХСН некалийсберегаю-щих диуретиков можно ожидать транзиторного повышения активности РААС, уровней АТ II и альдостерона, а также снижение содержания электролитов (калия и магния). Поэтому назначение АА в данной ситуации патофизиологически обосновано. Надо помнить, что у пациентов с ХСН концентрация калия в сыворотке крови может не соответствовать его содержанию в клетках из-за развития оксидативного стресса и последующего нарушения транспорта калия в ткани (дефекты в работе Na/K аденозинтри-фосфатазы) [70]. Отсюда у них даже при высокой калиемии (>7,0 ммоль/л) характерные клинические и электрокардиографические симптомы могут отсутствовать, поскольку истинное тканевое содержание калия (в эритроцитах крови) находится в пределах нормы. Высказывается мнение, что у пациентов с ХСН значение калия в плазме крови, на которое надо ориентироваться, следует сдвигать на 0,5 ммоль/л от верхнего предела лабораторной нормы (лучше поддерживать уровень калия в пределах 4,5– 5,5 ммоль/л) [71]. Перед назначением АА важно оценить концентрацию креатинина в плазме крови. Ее повышение на терапии эплереноном в исследовании EPHESUS было незначительным: на 0,06 мг/дл против 0,02 мг/дл на плацебо (р<0,001). При высоком уровне креатинина (≥2,5 мг/дл) от назначения АА следует воздержаться. На фоне приема АА необходимо контролировать скорость клубочковой фильтрации и не назначать АА при очень низкой ее величине (<30 мл/мин).
Заключение
К сожалению, назначение АА дополнительно к другим нейрогормональным модуляторам в России является редкостью. По данным Российского регистра РЕКОРД, признаки СН в стационаре у больных с ИМпST (n=246) наблюдались у 66,5% на фоне стандартной терапии [72]. В то же время в стационаре получали АА только 16,7% пациентов, а при выписке – 14% пациентов.
Игнорировать сегодня полученные в исследовании EPHESUS у пациентов после ИМ с СН обнадеживающие результаты невозможно, поскольку высокоселективный АА эплеренон, даже добавленный к стандартной терапии (ИАПФ/БРА и БАБ), позволяет существенно снижать риск серьезных сердечно-сосудистых осложнений, включая ВСС и общую смертность. Среди несомненных преимуществ эплеренона перед неселективными АА предыдущего поколения – лучшая переносимость и низкая частота нежелательных явлений, большая доказательная база (EPHESUS и EMPHASIS-HF n≈10 006). Очевидно, что в рутинной клинической практике больным с ОИМ, осложненным клинической СН или дисфункцией ЛЖ (ФВ ЛЖ менее 40%), начинать терапию эплереноном необходимо с первых дней (в исследовании EPHESUS с 3-х суток) после достижения гемодинамической стабильности (подтверждено международными рекомендациями и данными) в дозе 25 мг/сут с последующим переходом на поддерживающую дозу (50 мг/сут) и на длительный период, а сам эплеренон рассматривать в качестве эффективного средства вторичной профилактики.
Литература
1. Бокерия Л.А., Гудкова Р.Г. Сердечно-сосудистая хирургия. М.: Изд-во НЦССХ им. А.Н.Бакулева РАМН, 2010.
2. Goldberg RJ, Spencer FA, Yarzebski J et al. A 25-year perspective into the changing landscape of patients hospitalized with acute myocardial infarction (the Worcester Heart Attack Study). Am J Cardiol 2004; 94: 1373–8.
3. Solomon SD, Zelenkofske S, McMurray JJV et al. Sudden death in pa-tients with myocardial infarction and left ventricular dysfunction, heart failure, or both. N Eng J Med 2005; 325: 2581–8.
4. de Gevigney G, Ecochard R, Rabilloud M et al. Worsening of heart fail-ure during hospital course in myocardial infarction is a factor of poor prognosis. Apropos of a prospective cohort study of 2,507 patients hos-pitalized with myocardial infarction: PRIMA study. Ann Cardiol Angeiol (Paris) 2002; 51: 25–32.
5. Spencer FA, Meyer TE, Gore JM, Goldberg RJ. Heterogeneity in the man-agement and outcomes of patients with acute myocardial infarction complicated by heart failure. The National Registry of Myocardial In-farction. Circulation 2002; 105: 260–510.
6. Vaur L, Danchin N, Genes J et al. Epidemiology of myocardial infarction in France: therapeutic and prognostic implications of heart failure during the acute phase. Am Heart J 1999; 137: 4958.
7. Hellermann JP, Jacobsen SJ, Reeder G et al. Heart failure after my-ocardial infarction: prevalence of preserved left ventricular systolic function in the community. Am Heart J 2003; 145: 7428.
8. Steg PG, Dabbous OH, Feldman LJ et al. for the Global Registry of Acute Coronary Events (GRACE) Investigators. Determinants and prognostic impact of heart failure complicating acute coronary syndromes: obser-vations from the Global Registry of Acute Coronary Events (GRACE). Cir-culation 2004; 109: 494–9.
9. The CAPRICORN Investigators. Effect of carvedilol on outcome after myocardial infarction in patients with left-ventricular dysfunction: the CAPRICORN randomized trial. Lancet 2001; 357: 1385–90.
10. Архипов М.А., Бережинский И.В., Иващенко А.А. Ишемическое ремоделирование левого желудочка: методологические аспекты, вопросы диагностики и лечения. Под ред. Л.А.Бокерия и др. М., 2002.
11. Беленков Ю.Н. Ремоделирование левого желудочка: комплексный подход. Сердечная недостаточность 2002; 3 (4): 161–3.
12. Spinale FG, Coker ML, Heung LJ et al. A matrix metalloproteinase in-duction/activation system exists in the human left ventricular myocardi-um and is upregulated in heart failure. Circulation 2000; 102: 1944–9.
13. Mortensen RM, Williams GH. Aldosterone action in: DeGroot LJ, Jameson JL eds. Endocrinology, 4th edition. WB Saunders, Philadelphia, PA 2001: 1783–9.
14. McMachon E. Recent studies with eplerenone, a novel selective al-dosterone receptor antagonist. Curr Opin Pharmacol 2001; 1: 190–6.
15. Wang W. Chronic administration of aldosterone depresses barore-ceptor reflex function in the dog. Hypertension 1994; 24: 571–5.
16. Schmidt BMW, Montealegre A, Janson CP et al. Short term cardiovas-cular effects of aldosterone in healthy male volunteers. J Endocrinol Metab 1999; 84: 3528–33.
17. Booth E, Johnson JP, Stockand JD. Aldosterone. Adv Physiol Educ 2002; 26: 8–23.
18. Falkenstein E, Tillmann HC, Christ M et al. Multiple actions of steroid hormones – a focus on rapid, nongenomic effects. Pharmacol Rev 2000; 52: 513–56.
19. Silvestre J-S, Heymes C, Oubenaissa A et al. Activation of cardiac al-dosterone production in rat myocardial infarction: effect of angiotensin II receptor blockade and role in cardiac fibrosis. Circulation 1999; 99: 2694–701.
20. Danser A, Cha W. Cardioprotective effects of eplerenone in the rat heart: interaction with locally synthesized or blood-derived aldos-terone? The FASEB J 2006; 20: A342–A3.
21. Duprez KT, Bauwens FR, Buyzere ML et al. Influence of aeterial blood pressure and aldosterone on left ventricular hypertrophy in mod-erate essential hypertension. Am J Cardiol 1993; 71: 17A–20A.
22. Schunkert H, Hense HW, Danser J et al. Association between circulating components of the rennin-angiotensine-aldosterone system and left ventricular mass. Br Heart J 1997; 77: 24–31.
23. Sato A, Funder JW. High glucose stimulates aldosterone-induced hy-pertrophy via type I mineralocorticoid receptors in neonatal rat car-diomyocytes. Endocrinol 1996; 137: 4145–53.
24. Rocha R, Stier CT Jr, Kifor I et al. Aldosterone: a mediator of myocar-dial necrosis and renal arteriopathy. Endocrinol 2000; 141: 3871–8.
25. Weber KT, Brilla CG. Pathological hypertrophy and cardiac intersti-tium: fibrosis and renin-angiotensinaldosterone system. Circulation 1991; 83: 1849–65.
26. Brilla CG, Zhou G, Matsubara L, Weber KT. Collagen metabolism in cultured adult rat cardiac fibroblasts: response to angiotensin II and al-dosterone. J Moll Cell Cardiol 1994; 26: 809–20.
27. Rocha R, Rudolph AE, Frierdich GE et al. Aldosterone induces a vas-cular inflammatory phenotype in the rat heart. Am J Physiol Heart Circ Physiol 2002; 283: H1802–10.
28. Sun Y, Zhang J, Lu L et al. Aldosterone-induced inflammation in the rat heart. Role of oxidative stress. Am J Pathol 2002; 161: 1773–81.
|
29. Robert V, Heymes C, Silvestre JS et al. Angiotensin AT1-receptor sub-type as a cardiac target of aldosterone: role in aldosterone-salt induced fibrosis. Hypertension 1999; 33: 981–6.
30. Fullerton MJ, Funder JW. Aldosterone regulates collagen output of rat cardiac fibroblasts by up-
regulation of endothelin receptors. Endocrinol Soc Proc 1998; 93: 511–6.
31. Rude MK, Duhaney TAS, Kuster G et al. Aldosterone stimulates ma-trix metalloproteinases and reactive oxygen species in adult rat ven-tricular cardiomyocytes. Hypertention 2005; 46: 555–61.
32. Mano A, Tatsumi T, Shiraishi J et al. Aldosterone directly induces my-ocyte apoptosis through calcineurin-dependent pathways. Circulation 2004; 110: 317–23.
33. Duprez D, De Buyzere M, Rietzschel ER et al. Aldosterone and vascu-lar damage. Curr Hypertens
Rep 2000; 2: 327–34.
34. Mitchell BM, Smith AD, Webb RC, Dorrance AM. Aldosterone decreas-es endothelium-dependent relaxation by down-regulating GTP cyclo-hydrolase. Hypertension 2003; 42: 435 (P216). Abstract.
35. Sun Y, Zhang J, Lu L. Aldosterone-induced inflammation in the rat heart: role of oxidative stress. Am J
Pathol 2002; 161: 1773–81.
36. Rajagopalan S, Duquaine D, King S et al. Mineralocorticoid receptor antagonism in experimental atherosclerosis. Circulation 2002; 105: 2212–6.
37. Brown NJ, Kim KS, Chen YQ et al. Synergistic effect of adrenal steroids and angiotensin II on
plasminogen activator inhibitor-1 production. J Clin Endocrinol Metab 2000; 85: 336–44.
38. Brown NJ, Nakamura S, Ma L et al. Aldosterone modulates plas-minogen activator inhibitor-1 and glomerulosclerosis in vivo. Kidney Int 2000; 58:1219–27.
39. Сытник Н.В., Кокорин В.А., Люсов В.А. и др. Активность РААС и САС у больных в отдаленные
сроки после первичного инфаркта миокарда. Рос. кардиол. журн. 2009; 4: 17–22.
40. Palmer BR, Pilbrow AP, Frampton CM et al. Plasma aldosterone lev-els during hospitalization are predictive of survival post-myocardial in-farction. Eur Heart J 2008; 29: 2489–96.
41. Beygui F, Collet J-P, Benoliel J-J et al. High Plasma Aldosterone Levels on Admission Are Associated
With Death in Patients Presenting With Acute ST-Elevation Myocardial Infarction. Circulation 2006; 114:
2604–10.
42. Solomon SD, Zelenkofske S, McMurray JJV et al. Sudden death in pa-tients with myocardial infarction and left ventricular dysfunction, heart failure, or both. N Engl J Med 2005; 352: 2581–8.
43. The CONSENSUS Trial Study Group. Effects of enalapril on mortality in severe congestive heart failure. N Eng Med 1987; 316: 429–35.
44. Weber KT, Villarreal D. Aldosterone and antialdosterone therapy in congestive heart failure. Am J Cardiol 1993; 71: 3A–11A.
45. Georges B, Beguin C, Jadoul M. Spironolactone and congestive heart failure. Lancet 2000; 355: 1369–
70.
46. McManus F, McInnes GT and Connell JMC. Drug insight: eplerenone, a mineralocorticoid-receptor antagonist. Nature Clinical Practice: En-docrinology & Metabolism 2008; 4 (1): 44–52.
47. Staessen J, Lijnen P, Fagard R et al. Rise in plasma concentration of aldosterone during long-term
angiotensisn II suppression. J Endocrinol 1981; 91: 457–65.
48. Lee AFC, MacFadyen RJ, Struthers AD. Neurohormonal reactivation in heart failure patients on chronic ACE inhibitor therapy: a longitudi-nal study. Eur J Heart Fail 1999; 1: 401–6.
49. Sato A, Saruta T. Aldosterone escape during angiotensin-converting enzyme inhibitor therapy in
essential hypertensive patients with left ventricular hypertrophy. J Intern Med Res 2001; 29: 13–21.
|
50. Pitt B «Escape» of aldosterone production in patients with left ven-tricular dysfunction treated with an
angiotensin converting enzyme in-hibitor, implications for therapy. Cardiovasc Drug Ther 1995; 9: 145–9.
51. Tsutamoto T, Wada A, Maeda K et al. Effect of spironolactone on plasma brain natriuretic peptide and left ventricular remodeling in pa-tients with congestive heart failure. J Am Coll Cardiol 2001; 37: 1228–33.
52. deGasparo M, Joss U, Ramjoue et al. Three new epoxy-spironolac-tone derivates: characterization in vivo and in vitro. J Pharmacol Exp Ther 1987; 240: 650–56.
53. Weinberger MH, Roniker B, Krause SL et al. Eplerenone, a selective al-dosterone blocker, in mild-to-
moderate hypertension. Am J Hypertens 2002; 15: 709–16.
54. Pitt B, Remme W, Zannad F et al. Eplerenone, a Selective Aldosterone Blocker, in Patients with Left
Ventricular Dysfunction after Myocardial Infarction. N Engl J Med 2003; 348: 1309–21.
55. Pitt B, Zannand F, Remme W et al. The effect of spirinilactone on morbidity and mortality in patients with severe heart failure. N Engl J Med 1999; 341: 709–17.
56. ACC/AHA 2005 Guideline Update for the Diagnosis and Management of Chronic Heart Failure in the
Adult. J Am Coll Cardiol 2005; 46: 1–82.
57. Pitt B, White H, Nicolau J et al. Eplerenone Reduces Mortality 30 Days After Randomization Following Acute Myocardial Infarction in Patients With Left Ventricular Systolic Dysfunction and Heart Failure. J Am Coll Cardiol 2005; 46: 425–31.
58. Pitt B, Gheorghiade M, Zannad F et al. On behalf of the EPHESUS In-vestigators. Evaluation of eplerenone in the subgroup of EPHESUS pa-tients with baseline left ventricular ejection fraction < or
=30%. Eur J Heart Fail 2006; 8 (3): 295–301.
59. Gheorghiade M, Khan S, Blair JEA et al. The effect of eplerenone on length of stay and total days of heart failure hospitalization after my-ocardial infarction in patients with left ventricular systolic dysfunction. Am Heart J 2009; 158: 437–43.
60. Suzuki G, Morita H, Mishima T et al. Effects of long-term monothera-py with eplerenone, a novel aldosterone blocker, on progression of left ventricular dysfunction and remodeling in dogs with heart failure. Cir-culation 2002; 106: 2967–72.
61. Hayashi M, Tsutamoto T, Wada A et al. Immediate administration of mineralocorticoid receptor antagonist spironolactone prevents post-infarct left ventricular remodeling associated with suppression of my-ocardial collagen synthesis in patients with first anterior acute myocar-dial infarction. Circulation 2003;
107: 2559–65.
62. Zannad F, Ketelsiegers JM, Schiffrin EL et al. The effect of eplerenone on markers of cardiac fibrosis:
insights from EPHESUS. J Am Coll Cardiol 2004; 43 (Suppl. A): A200.
63. Ketelsiegers JM, Zannad F, Schiffrin EL et al. The effect of eplerenone on the cytokine osteopontin in post-AMI heart failure: an EPHESUS sub-study Eur Heart J 2004; 25 (Suppl.): 485.
64. Trueblood NA, Xie Z, Cormmunal C et al. Exaggerated left ventricular dilation and reduced collagen deposition after myocardial infarction in mice lacking osteopontin. Circ Res 2001; 88: 1080–7.
65. Rocha R, Rudolph AE, Frierdich GE et al. Aldosterone induces a vas-cular inflammatory phenotype in
the rat heart. Am J Physiol. Heart Circ Physiol 2002; 283: H1802–10.
66. Ramires FJA, Mansur A, Coelho O et al. Effect of spironolactone on ventricular arrhythmias in congestive heart failure secondary to idio-pathic dilated or to ischemic cardiomyopathy. Am J Cardiol
2000; 85: 1207–11.
|
67. Delyani JA, Robincon EL, Rudolph AE. Effect of a selective aldosterone receptor antagonist in myocardial infarction. Am J Physiol Heart Circ Physiol 2001; 281: H647–54.
68. Zannad F, McMurray JJ, Krum H et al. Eplerenone in Patients with Systolic Heart Failure and Mild
Symptoms. N Engl J Med 2011; 364 (1):11—21.
69. Pitt B, Bakris G, Ruilope LM et al. Serum Potassium and Clinical in the Eplerenon Post-Acute
Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS). Circulation 2008; 118: 1643–
50.
70. Macdonald JE, Struthers AD. What is the optimal serum potassium level in cardiovascular patients? J Am Coll Cardiol 2004; 43: 155–61.
71. Delgado-Almeida A, Delgado-Leon C. Changes in plasma ionized calcium and RBC K content in severe hyperkalemia: new electrocardio-graphic concept. Circulation 2006; 114 (Suppl. II): II-466.
72. Эрлих А.Д., Грацианский Н.А. Регистр острых коронарных синдромов РЕКОРД. Характеристика
|
больных и лечение до выписки из стационара. Кардиология. 2009; 7–8: 4–12.




Комментировать