

Воспалительные поражения миокарда. Острая, подострая и хроническая фаза вирусного миокардита и ее характеристики. Изучение количественных параметров и особенностей функционального состояния основных субпопуляций лимфоцитов периферической крови больных миокардитом с различной степенью выраженности гемодинамических нарушений. Результаты проведенных исследований.
Палеев Н.Р., Палеев Ф.Н., Санина Н.П., Макарков А.И., Москалец О.В., Островский Е.И., Хишова Н.Н.
Воспалительные поражения миокарда представляют собой гетерогенную группу заболеваний, различающихся как этиологией (вирусные, бактериальные, грибковые инфекции, протозойные инвазии и гельминтозы, действие токсинов, физических и химических факторов, аутоиммунные механизмы и реакции гиперчувствительности), так и уникальным для каждой фазы воспалительного процесса сочетанием патогенетических механизмов [1, 2].
Принято считать, что воспаление миокарда лежит в основе около 10% случаев сердечной недостаточности [3]. Истинную заболеваемость миокардитами оценить сложно из-за мало- или бессимптомного течения заболевания в значительном числе случаев, однако минимальная клиническая манифестация острой фазы миокардита не исключает в последующем дилатации полостей сердца с развитием картины хронической сердечной недостаточности [4]. По результатам патолого-анатомических исследований, частота случаев миокардита ; колеблется в диапазоне от 0,12 до 12% [5].
В основе воспалительного процесса при миокарде чаще всего лежат вирусная инфекция и вирус-индуцированные иммунные реакции [6, 7]. В качестве этиологического фактора при миокардите обычно указывают энтеровирусы, и прежде всего вирус Коксаки B3, однако в последние годы пристальное внимание стали уделять парвовирусу B19 и вирусам герпеса [8, 9]. Недавно была сформулирована концепция о фазовом течении вирус-индуцированного миокардита, где отмечены три фазы – острая, подострая и хроническая – и выявлены ключевые механизмы патогенеза для каждой из фаз заболевания [4, 8].
Ключевым событием острой фазы вирусного миокардита является внедрение вирусной частицы, обладающей тропизмом к сократительному миокарду, в клетку-мишень, что приводит к ее повреждению или гибели. Прямое цитопатогенное действие вируса является ведущим механизмом повреждения миокарда в острой фазе заболевания. Кроме того, включаются неспецифические механизмы противовирусной защиты, направленные на элиминацию инфицированных кардиомиоцитов и представленные естественными киллерными (natural killer – NK) и NKT-клетками и макрофагами. Макрофаги служат источником провоспалительных цитокинов, а NK-клетки обеспечивают перфорин и гранзим-опосредованный лизис клеток сократительного миокарда. Продолжительность острой фазы вирусного миокардита составляет всего несколько дней, а выраженность клинических проявлений варьирует от бессимптомного до остро протекающего заболевания. Следует отметить, что эффективная элиминация инфекционного патогена благодаря включению механизмов иммунной защиты при остром течении заболевания обеспечивает благоприятный долговременный прогноз: 93% пациентов, переживших острую фазу заболевания, выживали в течение 11 лет, по сравнению с 45% больных с острым миокардитом [10].
Таким образом, острая фаза вирусного миокардита имеет следующие характеристики:
- высокий удельный вес цитопатогенного действия вирусов в повреждении миокарда;
• участие неспецифических механизмов противовирусной защиты, реализуемых макрофагами и NK-клетками;
• реакция иммунной системы адекватна повреждающему фактору, нет признаков иммунопатологии;
• в основе гибели кардиомиоцитов лежат механизмы некроза.
В тех случаях, когда неспецифические механизмы противовирусной защиты не могут обеспечить элиминацию вирусного генома, заболевание переходит в подострую фазу, основным содержанием которой является включение механизмов специфической иммунной защиты. Активированные макрофаги и другие клетки иммунной системы посредством продукции хемокинов привлекают в очаг воспаления Т- и В-лимфоциты, которые, в свою очередь, реализуют механизмы клеточно-опосредованного цитолиза, а также обеспечивают выработку противовирусных антител. Специфические иммунные механизмы направлены как против вирусных частиц, так и против антигенов собственных клеток, обладающих сходством с вирусными детерминантами, либо ставших доступными для иммунной атаки вследствие попадания во внеклеточное пространство или усиления иммуногенности по причине изменения нативной антигенной структуры.
Массивная продукция провоспалительных цитокинов активированными макрофагами и эффекторными
Т-лимфоцитами запускает механизм апоптоза кардиомиоцитов, что усугубляет снижение сократимости миокарда.
Специфический иммунный ответ обеспечивает максимальную противовирусную защиту, однако потенциально способен приобрести черты аутоиммунного процесса. Включение механизмов самоограничения, прежде всего за счет активации «регуляторных» Т-клеток (Treg), а также поддержания баланса субпопуляций хелперных Т-лимфоцитов, обеспечивает физиологическое затухание иммунных реакций после элиминации вирусного патогена. Продолжительность подострой фазы вирусного миокардита составляет от нескольких недель до нескольких месяцев.
Таким образом, подострая фаза вирусного миокардита имеет следующие характеристики:
- подострый вирусный миокардит развивается в том случае, если механизмы неспецифической противовирусной защиты, в т. ч. макрофаги и NK-клетки, не смогли обеспечить элиминацию вирусного генома;
• в основе лежат специфические механизмы клеточного и гуморального иммунитета;
• велик удельный вес иммунного повреждения кардиомиоцитов;
• иммунный ответ носит физиологический характер, в целом адекватен степени выраженности нарушения антигенного гомеостаза;
• как некроз, так и механизмы апоптоза лежат в основе гибели кардиомиоцитов.
Сущностью хронической фазы вирусного миокардита является трансформация заболевания в дилатацию. В основе патологического процесса лежит персистирующее воспаление миокарда вследствие неадекватности иммунного ответа. Неадекватность иммунного ответа заключается в неспособности элиминировать вирусный патоген либо в несостоятельности механизмов самоограничения иммунных процессов [11]. Соответственно, вирус может присутствовать в миокарде, поддерживая воспалительную реакцию, либо повреждение миокарда приобретает аутоиммунный характер и продолжается после элиминации вирусного патогена. Воспаление миокарда опосредовано специфическими иммунными механизмами и присутствует в течение продолжительного времени, приводя к утрате сократительного миокарда, дилатации полостей сердца и увеличению доли соединительнотканного компонента вследствие избыточной продукции профиброгенных цитокинов. Ведущую роль в гибели кардиомиоцитов играют механизмы апоптоза. Изменения в миокарде носят необратимый характер [11, 12].
Важнейшими характеристиками хронической фазы миокардита являются следующие:
- специфические иммунные механизмы лежат в основе прогрессирующего повреждения миокарда;
• иммунный ответ носит патологический характер, что выражается в неспособности иммунной системы к элиминации вируса или в несостоятельности механизмов самоограничения иммунного ответа;
• вирусный геном может присутствовать в клетках сердца, либо заболевание приобретает аутоиммунный характер. Это необходимо учитывать при выборе врачебной тактики – назначении противовирусных препаратов или иммуносупрессивной терапии [13];
• апоптоз кардиомиоцитов становится ведущим механизмом утраты сократительного миокарда;
• ремоделирование миокарда носит, как правило, необратимый характер.
Приведенная выше концепция носит общий характер. В частности, она не учитывает тропность вирусов к различным клеткам миокарда – кардиомиоцитам (энтеровирусы), эндотелию сосудов (парвовирусы, вирусы герпеса) или фибробластам, которая определяет своеобразие клинической картины заболевания, течение и прогноз [14, 15]. Тем не менее, эта концепция хорошо иллюстрирует ведущую роль иммунных механизмов при воспалительных поражениях миокарда [1, 16].
Целью нашего исследования явилось изучение количественных параметров и особенностей функционального состояния основных субпопуляций лимфоцитов периферической крови больных миокардитом с различной степенью выраженности гемодинамических нарушений. В работе приняли участие 30 больных инфекционно-иммунным миокардитом, у 13 из которых отмечались выраженные симптомы сердечной недостаточности (II–IV функциональный класс по классификации Нью-Йоркской кардиологической ассоциации – NYHA). У остальных 17 пациентов признаки сердечной недостаточности отсутствовали или были минимальными (I функциональный класс по NYHA). Контрольную группу составили 10 практически здоровых людей. Показатели гемодинамики исследовали методом двухмерной и допплер-эхокардиографии. Анализ спектра трансмитрального диастолического потока использовали для оценки диастолической функции сердца. Исследование популяционного и субпопуляционного состава и активационных маркеров лимфоцитов периферической крови проводили методом четырехцветной лазерной проточной цитометрии c использованием прибора FACSCalibur и моноклональных антител (Becton Dickinson, США). Статистическую обработку результатов выполняли с помощью программы PASW Statistics 18. Для определения статистической обоснованности различия исследуемых групп применяли непараметрический U-критерий Манна–Уитни. Для оценки характера, силы и достоверности связи отдельных показателей использовали коэффициент линейной корреляции Пирсона. В таблицах 1 и 2 представлены средняя величина (M) и ее стандартная ошибка (m) для каждого показателя. На рисунке 1 отражены средние величины изучаемых показателей.
В таблице 1 представлены результаты эхокардиографического обследования. У больных миокардитом с выраженной сердечной недостаточностью были отмечены признаки систолической и диастолической дисфункции левого желудочка и значительная дилатация левых и правых отделов сердца. Напротив, у пациентов с минимальными признаками сердечной недостаточности отсутствовали значимые изменения размеров и полостей сердца и признаки диастолической дисфункции, а величина ФВ хотя и была ниже, чем в контрольной группе, оставалась в пределах нормальных значений.
Количественные характеристики основных субпопуляций Т-лимфоцитов у больных миокардитом существенно не отличались от показателей контрольной группы (табл. 2). Следует, однако, отметить наличие корреляционной связи (r=0,43; p<0,01) между числом CD3+CD4+ лимфоцитов и временем замедления раннего диастолического потока (показатель DT, позволяющий выявить диастолическую дисфункцию левого желудочка). При этом связь оказалась сильной в группе больных с выраженными симптомами сердечной недостаточности (r=0,74; p<0,05) и отсутствовала у пациентов с минимальной выраженностью сердечной недостаточности и в контрольной группе. Кроме того, мы выявили сильную отрицательную связь между числом цитотоксических CD3+CD8+ лимфоцитов и величиной фракции выброса левого желудочка в группе больных с выраженными симптомами сердечной недостаточности (r=–0,80; p<0,01), но не у пациентов с минимальными симптомами сердечной недостаточности или в контрольной группе. Полученные результаты указывают на вероятную роль иммунных механизмов в развитии систолической и диастолической дисфункции левого желудочка при миокардите.
Большого внимания заслуживает сообщение о двукратном снижении числа CD3+CD16/56+ лимфоцитов в крови больных миокардитом (с 13,9% до 5,3%, p<0,01). Клетки, коэкспрессирующие поверхностные маркеры CD3 и CD16 и/или CD56, принадлежат к уникальной субпопуляции Т-лимфоцитов, получившей название NKT-клетки и обладающей свойствами Т- и NK-лимфоцитов [17]. Реализуя функции, свойственные NK-клеткам, они одновременно являются активными продуцентами цитокинов, что объясняет их участие в регуляции про- и противовоспалительных иммунных реакций, а также центральную роль в противовирусном иммунитете [18].
NKT-клетки представляют собой гетерогенную группу, включающую инвариантные NKT-клетки (тип I), NKT-клетки II типа и NKT-подобные клетки [19]. NKT-клетки распознают липидные и низкомолекулярные нелипидные антигены в комплексе с альтернативными антигенпрезентирующими молекулами CD1a, CD1b, CD1c и CD1d, а также MR1. Они принадлежат к субпопуляциям CD4+, CD8+ или CD4–CD8-лимфоцитов. Часть NKT-клеток экспрессируют поверхностный антиген CD161, являющийся маркером NK- и Th17-лимфоцитов. NKT-клетки способны секретировать цитокины, характерные как для Th1-, так и Th2-субпопуляций Т-хелперов, интерлейкина (ИЛ) 17А, экспрессируют рецептор ИЛ-17RB [19, 20].
Широта цитокинового спектра NKT-клеток объясняет многообразие их эффектов при воспалительных поражениях миокарда. Активация NKT-клеток является мощным механизмом противоинфекционного иммунитета. В частности, при заболевании, вызванном вирусом Коксаки B3, усиливается экспрессия CD1d на макрофагах, дендритных клетках и Т-лимфоцитах и индуцируется секреция иммунными клетками интерферона (ИФН-γ) [21]. Инфицирование вирусом энцефаломиокардита сопровождается CD1d-зависимой активацией NKT-лимфоцитов, за которой следует активация Т- и NK-клеток, выработка противовирусных факторов, в т. ч. ИФН-γ и ИФН-α, и, как результат, подавление вирусной репликации [22]. ИФН-γ-зависимым образом NKT-клетки обеспечивают усиление противоинфекционной защиты при бактериальном миокардите, вызванном Borrelia burgdorferi: ИФН-γ облегчает распознавание и фагоцитоз микробов макрофагами, одновременно увеличивая экспрессию CD1d-молекул на антиген-презентирующих клетках и тем самым замыкая цикл положительной обратной связи, приводящий к дополнительной активации NKT-клеток [23]. С другой стороны, ряд инфекционных агентов, в т. ч. обладающих кардиотропизмом (лейшмания, цитомегаловирус, вирус простого герпеса), угнетают экспрессию молекул CD1. Поскольку иммунные реакции, связанные с CD1-опосредованной презентацией бактериальных и вирусных антигенов, способствуют защите организма от инфекции, снижение экспрессии CD1 создает условия для персистенции патогена. Помимо нарушения процессов презентации антигена подавление иммунных реакций NKT-клетками может быть опосредовано активацией Treg-лимфоцитов. Показана способность низкопатогенного варианта вируса Коксаки B3 активировать регуляторные CD4+CD25+ клетки, не вызывая одновременного усиления экспрессии CD1d-антигена. NKT-лимфоциты секретируют значительные количества ИЛ-10 и трансформирующего фактора роста β, что приводит к подавлению экспрессии вспомогательных активационных молекул CD40, CD80 и CD86 на антиген-презентирующих клетках и ограничению иммунных реакций [11, 24]. Таким образом, роль NKT-лимфоцитов при миокардите может состоять как в усилении цитотоксичности CD8+ лимфоцитов в отношении клеток миокарда, так и в ограничении избыточных иммунных реакций и предотвращении аутоиммунизации. Последний механизм, однако, создает предпосылки для персистенции инфекционного патогена в организме больного [25].
Обнаруженное нами снижение числа CD3+CD16/56+ лимфоцитов при миокардите, выявляемое у пациентов с различной степенью сердечной недостаточности, важно в контексте обсуждения баланса противовирусных, цитотоксических и аутоиммунных механизмов при воспалении миокарда и свидетельствует, вероятно, о нарушении механизмов противовирусной защиты. Это предположение косвенно подтверждается положительной корреляцией между количеством CD3+CD16/56+ и цитотоксических CD3+CD8+ лимфоцитов (r=0,50; p<0,01 у больных миокардитом и r=0,78; p<0,01 в контрольной группе). Ранее мы сообщали об отрицательной корреляционной связи между числом CD3+CD16/56+ лимфоцитов и сывороточными концентрациями противовоспалительных цитокинов ИЛ-4 и ИЛ-10 у здоровых доноров [26]. Для проверки данной гипотезы необходимы дальнейшие иммунологические и микробиологические исследования, в т. ч. изучение цитокинового профиля CD3+CD16/56+ лимфоцитов при миокардите, параметров вирусной репликации, уточнение фенотипа NKT-клеток (CD3+CD16+CD56+, CD3+CD16–CD56+ или CD3+CD16+CD56–), а также их принадлежности к CD4+, CD8+ или CD4–CD8-субпопуляциям Т-лимфоцитов.
При исследовании последовательности экспрессии активационных маркеров лимфоцитами больных острым миокардитом через 2 нед. от начала заболевания отмечался прирост экспрессии всех исследованных маркеров активации, однако наиболее ранним и значительным иммунологическим признаком острого миокардита являлся прирост доли СD25+ клеток [27]. Через 3 нед. от начала миокардита сохранялась повышенная экспрессия антигена CD25, однако самой существенной иммунологической особенностью этого периода заболевания становился прирост числа CD71+ лимфоцитов. К концу месяца отмечалась нормализация экспрессии ранних активационных маркеров лимфоцитов, однако доля клеток, несущих антиген HLD-DR, была повышенной. Наконец, примерно через 2 мес. от начала заболевания экспрессия активационных маркеров лимфоцитами больных миокардитом практически соответствовала уровню здоровых доноров. Таким образом, для больных миокардитом была характерна специфическая последовательность экспрессии маркеров активации лимфоцитов CD25 → CD71 → HLA-DR, что соответствует нормальному течению и отражает гармоничный характер активации иммунной системы.
Важно отметить, что прирост числа Т-лимфоцитов, экспрессирующих ранний активационный маркер CD25, мы обнаружили в группе больных с минимальными симптомами сердечной недостаточности. Напротив, у больных с выраженными признаками сердечной недостаточности доля Т-клеток, экспрессирующих маркеры активации CD25 и HLA-DR, не отличалась от показателей контрольной группы, зато количество активированных «не Т-лимфоцитов» с фенотипом CD3–HLA-DR+, относящихся к популяциям B- и NK-клеток, было на 57% выше, чем у здоровых людей. Положительная корреляционная связь между числом CD3–HLA-DR+ и CD19+ В-лимфоцитов у больных миокардитом (r=0,43; p<0,05) указывает на активацию гуморальных иммунных механизмов у пациентов с выраженными симптомами сердечной недостаточности.
Представляет интерес комплексная оценка экспрессии активационных маркеров лимфоцитов и антигена CD95, отражающего готовность лимфоцита к реализации программы апоптотической гибели. Ранее мы сообщали о нарушении механизмов активационного апоптоза лимфоцитов у больных миокардитом с неблагоприятным течением заболевания [28]. В настоящем исследовании мы обнаружили значительный прирост числа CD3+CD95+-лимфоцитов периферической крови при миокардите, особенно в группе больных с выраженными признаками сердечной недостаточности.
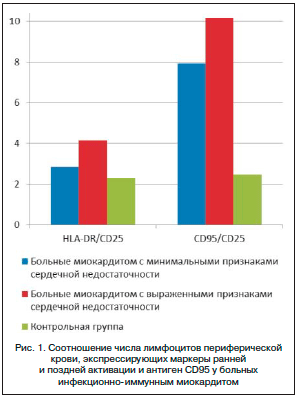
Для оценки последовательности активационных событий и способности лимфоидной клетки к переходу от активации к активационной гибели мы рассчитывали коэффициенты HLA-DR/CD25 и CD95/CD25. Как видно из рисунка 1, у больных миокардитом преобладают лимфоидные клетки, несущие признаки поздней активации и готовности к реализации программы CD95-опосредованного апоптоза, причем указанные сдвиги наиболее заметны в группе пациентов с выраженными симптомами сердечной недостаточности. Величина индекса HLA-DR/CD25 составила 2,58 у пациентов с минимальными признаками сердечной недостаточностью и 4,14 – у больных с выраженными симптомами (величина показателя в контрольной группе составила 2,29). Соотношение CD95/CD25 составило 7,93 у больных с минимальными симптомами и 10,16 – у пациентов с выраженной сердечной недостаточностью (в контрольной группе показатель был равен 2,47). Следует подчеркнуть, что экспрессия антигена CD95 не означает, что лимфоидная клетка неизбежно реализует апоптотическую программу. Напротив, нарушения в системе CD95-CD95L приводят к накоплению в крови аутореактивных лимфоцитов, что является ключевым патогенетическим механизмом аутоиммунного повреждения [29]. Для ответа на вопрос об «эффективности» апоптотического механизма необходимо исследование маркеров «состоявшегося» апоптоза лимфоцитов у больных миокардитом.
Таким образом, особенности иммунного статуса больных инфекционно-иммунным миокардитом необходимо рассматривать в контексте баланса реакций противовирусной защиты и аутоиммунного повреждения сердечной мышцы. При миокардите выявлено значительное снижение числа NKT-лимфоцитов, обладающих противовирусной и цитотоксической активностью. Т-лимфоциты больных миокардитом с минимальными проявлениями сердечной недостаточности характеризовались признаками «ранней» активации, выявляемой по повышенной экспрессии маркера CD25. Напротив, в крови больных с выраженными симптомами сердечной недостаточностью было отмечено увеличение числа активированных «не Т–клеток», связанное, вероятно, со стимуляцией гуморальных иммунных реакций. Наконец, иммунный статус больных миокардитом характеризовался преобладанием лимфоцитов, несущих признаки «поздней» активации и готовности к реализации апоптотической программы, особенно в группе с выраженными симптомами сердечной недостаточности. Последнее наблюдение указывает на вероятное нарушение механизмов активационного апоптоза лимфоцитов при инфекционно-иммунном миокардите.
Представленные выше сведения иллюстрируют важную роль нарушений противоинфекционного иммунитета и механизмов самоограничения иммунных реакций в возникновении и прогрессировании поражений миокарда воспалительной природы. Прогресс в разработке новых, современных методов диагностики и лечения миокардитов возможен только при условии комплексного изучения особенностей врожденного и адаптивного иммунитета при этом заболевании. Большой интерес представляет исследование патогенетических и компенсаторно-приспособительных иммунных механизмов, в т. ч. реализуемых недавно охарактеризованными Th17- и Th22- субпопуляциями Т-лимфоцитов [16, 30–32]. Понимание тонких механизмов патогенеза воспаления сердечной мышцы является необходимым условием создания «точечных» инструментов фармакологического воздействия, которые позволят нам приблизиться к решению проблемы профилактики, диагностики и лечения миокардитов.
Таким образом, особенности иммунного статуса больных инфекционно-иммунным миокардитом необходимо рассматривать в контексте баланса реакций противовирусной защиты и аутоиммунного повреждения сердечной мышцы. При миокардите выявлено значительное снижение числа NKT-лимфоцитов, обладающих противовирусной и цитотоксической активностью. Т-лимфоциты больных миокардитом с минимальными проявлениями сердечной недостаточности характеризовались признаками «ранней» активации, выявляемой по повышенной экспрессии маркера CD25. Напротив, в крови больных с выраженными симптомами сердечной недостаточностью было отмечено увеличение числа активированных «не Т–клеток», связанное, вероятно, со стимуляцией гуморальных иммунных реакций. Наконец, иммунный статус больных миокардитом характеризовался преобладанием лимфоцитов, несущих признаки «поздней» активации и готовности к реализации апоптотической программы, особенно в группе с выраженными симптомами сердечной недостаточности. Последнее наблюдение указывает на вероятное нарушение механизмов активационного апоптоза лимфоцитов при инфекционно-иммунном миокардите.
Литература
- Палеев Н.Р., Палеев Ф.Н., Санина Н.П. Миокардиты // Альманах клинической медицины. 2004. № 7. С. 118–126.
- Палеев Н.Р., Палеев Ф.Н. Некоронарогенные заболевания миокарда и их классификация // Рос. кардиол. журнал. 2009. № 3. С. 5–9.
- Yancy C.W., Jessup M., Bozkurt B. et al. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines // J. Am. Coll. Cardiol. 2013. Vol. 62. P. e147–239.
- Dennert R., Crijns H. J., Heymans S. Acute viral myocarditis // Eur. Heart. J. 2008. Vol. 29. P. 2073–2082.
5. Blauwet L.A., Cooper L.T. Myocarditis // Prog. Cardiovasc. Dis. 2010. Vol. 52. P. 274–288. - Палеев Н.Р., Палеев Ф.Н. Иммунопатология миокардитов // Креативная кардиология. 2007. № 1–2. С. 46–55.
- Schultheiss H.P., Kühl U., Cooper L.T. The management of myocarditis // Eur. Heart J. 2011. Vol. 32. P. 2616–2625.
8. Kindermann I., Barth C, Mahfoud F. et al. Update on Myocarditis // J. Am. Coll. Cardiol. 2012. Vol. 59. P. 779–792.
9. Qian Q., Xiong S., Xu W. Manipulating intestinal immunity and microflora: an alternative solution to viral myocarditis? // Future Microbiol. 2012. Vol. 7. P. 1207–1216. - McCarthy R.E. 3rd, Boehmer J.P., Hruban R.H. et al. Long-term outcome of fulminant myocarditis as compared with acute (nonfulminant) myocarditis // N. Engl. J. Med. 2000. Vol. 342. P. 690–695.
- Huber S.A., Feldman A.M., Sartini D. Coxsackievirus B3 induces T regulatory cells, which inhibit cardiomyopathy in tumor necrosis factor-alpha transgenic mice // Circ. Res. 2006. Vol. 99. P. 1109–1116.
- Макарков А.И., Салмаси Ж.М., Санина Н.П. Апоптоз и сердечная недостаточность // Сердечная недостаточность. 2003. № 6. С. 312–314.
- Frustaci A., Russo M. A., Chimenti C. Randomized study on the efficacy of immunosuppressive therapy in patients with virus-negative inflammatory cardiomyopathy: the TIMIC study // Eur. Heart J. 2009. Vol. 30. P. 1995–2002.
- Bultmann B. D., Klingel K., Sotlar K. et al. Fatal parvovirus B19-associated myocarditis clinically mimicking ischemic heart disease: an endothelial cell-mediated disease // Hum. Pathol. 2003. Vol. 34. P. 92–95.
15. Krueger G. R. F., Rojo J., Buja L. M., et al. Human herpesvirus-6 (HHV-6) is a possible cardiac pathogen: an immunihistological and ultrastructural study // Hosp. Gen. 2008. Vol. 71. P. 187–191. - Салмаси Ж. М., Санина Н. П., Макарков А. И. и др. Роль Тh17-опосредованных механизмов иммунорегуляции в патогенезе воспалительных поражений миокарда // Российский иммунологический журнал. 2012. № 3. С. 211-222.
- Mittag A., Lenz D., Gerstner A.O. et al. Polychromatic (eight-color) slide-based cytometry for the phenotyping of leukocyte, NK, and NKT subsets // Cytometry A. 2005. Vol. 65. P. 103-115.
- Никитин В. Ю., Сухина И. А., Цыган В. Н., Гусев Д. А. Иммунологическая характеристика стадий хронического Гепатита С и оценка факторов иммунной системы как прогностических критериев течения заболевания // Журнал инфектологии. 2009. № 1. С. 30-40.
- Simoni Y., Diana J., Ghazarian L. et al. Therapeutic manipulation of natural killer (NK) T cells in autoimmunity: are we close to reality? // Clin. Exp. Immunol. 2013. Vol. 171. P. 8-19.
- Новиков Д. К. Разнообразие путей распознавания антигенов и развития иммунного ответа, роль CD1 молекул // Иммунопатология, аллергология, инфектология. 2001. № 1. С. 5-12.
- Huber S. A. CD1d expression on hemopoietic cells promotes CD4+ Th1 response in coxsackievirus B3 induced myocarditis // Virology. 2006. Vol. 352. P. 226-236.
- Ilyinskii P. O., Wang R., Balk S. P., Exley M. A. CD1d mediates T-cell-dependent resistance to secondary infection with encephalomyocarditis virus (EMCV) in vitro and immune response to EMCV infection in vivo // J. Virol. 2006. Vol. 80. P. 7146-7158.
- Olson C. M. Jr, Bates T. C., Izadi H. et al., Local production of IFN-gamma by invariant NKT cells modulates acute Lyme carditis // J. Immunol. 2009. Vol. 182. P. 3728-3734.
- Toda A., Piccirillo C. A. Development and function of naturally occurring CD4+CD25+regulatory T cells // J. Leukoc. Biol. 2006. Vol. 80. P. 458–470.
- Liu W., Huber S. A. Cross-talk between cd1d-restricted nkt cells and γδ cells in t regulatory cell response // Virol. J. 2011. Vol. 8. P. 32.
- Санина Н. П., Хишова Н. Н., Москалец О. В., Макарков А. И. Характеристика CD3+CD16+CD56+ субпопуляции Т-лимфоцитов у больных инфекционно-иммунным миокардитом // Тезисы докладов III Евразийского конгресса кардиологов. Москва. 2014. С. 112.
- Poryadin G. V., Sanina N. P., Makarkov A. I. et al. Dynamic Characteristic of Surface Phenotype of Peripheral Blood Lymphocytes in Myocarditis Patients // Russ. J. Immunol. 1999. Vol. 4. P. 165-170.
- Porjadin G. V., Sanina N. P., Makarkov A. I. et al. Effectiveness of apoptosis of lymphocytes as a prognostic index in patients with myocarditis // Pathophysiology. 1998. Vol. 5(Suppl.). P. 43.
- Randhawa S. R., Chahine B. G., Lowery-Nordberg M. et al. Underexpression and overexpression of Fas and Fas ligand: a double-edged sword // Ann. Allergy Asthma Immunol. 2010. Vol. 104. P. 286-292.
- Baldeviano G. C., Barin J. G., Talor M. V. et al. Interleukin-17A Is Dispensable for Myocarditis but Essential for the Progression to Dilated Cardiomyopathy // Circ. Res. 2010. Vol. 106. P. 1646-1655.
- Camporeale A., Marino F., Papageorgiou A. et al. STAT3 activity is necessary and sufficient for the development of immune-mediated myocarditis in mice and promotes progression to dilated cardiomyopathy // EMBO Mol. Med. 2013. Vol. 5. P. 572-590.
- Kong Q., Wu W., Yang F. et al. Increased expressions of IL-22 and Th22 cells in the coxsackievirus B3-Induced mice acute viral myocarditis // Virol. J. 2012. Vol. 9. P. 232.




Комментировать