Общее описание. Генетика ПОФ и семейные исследования. Молекулярная генетика. Диагностика и лечение.
Антелава О.А., Никишина И.П., Гусева И.А., Мякоткин В.А., Хелковская-Сергеева А.Н., Раденска-Лоповок С.Г., Насонов Е.Л.
Прогрессирующая оссифицирующая фибродисплазия (ПОФ) (от лат. fibro – волокно, dis – расстройство, нарушение, plasis – строение, структура и os, ossis – кость, facio – делать, окостенение) – редкое аутосомно-доминантное заболевание, характеризующееся прогрессирующим гетеротопическим истинным окостенением мягких тканей скелета (соединительнотканных прослоек в толще мышц, фасций, апоневрозов, сухожилий) и ассоциирующееся с множественными врожденными скелетными аномалиями. В МКБ-10 ПОФ представлена отдельной рубрикой – M 61.1.
ПОФ – это редкая нозологическая форма, характеризующаяся рецидивирующими болезненными эпизодами припухлости мягких тканей, ведущими к гетеротопической оссификации через стадию эндохондрального процесса, т. е. превращением соединительной ткани межмышечных прослоек, сухожилий и связочного аппарата в костную ткань [1, 2].
В литературе это заболевание можно встретить под названиями: параоссальная гетеротопическая оссификация, болезнь Мюнхмайера, «болезнь второго скелета», опухоль Стернера [3, 4]. Наиболее часто употреблявшееся ранее название – «оссифицирующий миозит» («превращение мышцы в кость») в настоящее время считается некорректным, поскольку не вполне отражает патогенетические и патоморфологические основы болезни. Общепризнанным научным термином, обозначающий данную патологию, является термин «прогрессирующая оссифицирующая фибродисплазия»;, что указывает на вовлечение в процесс окостенения соединительнотканных структур вследствие особого характера воспалительных процессов в сухожилиях, связках, фасциях, апоневрозах и мышцах, заканчивающихся окостенением. Расовой, половой и географической предрасположенности к ПОФ не обнаружено [5].
Интерес к этому заболеванию существует с давних времен. Первое упоминание о ПОФ относится к 1648 г., когда Patin описал «окостеневшую» пациентку [6]. В 1996 г. R. Smith et al. проанализировали клиническую картину ПОФ у 28 больных, которых они наблюдали в течение 24 лет. При этом оссификация скелетных мышц начиналась в первые месяцы или годы от рождения с мышц шеи, верхних отделов спины, мышц, окружающих крупные суставы, с последующим распространением на бедра, плечи; поражались также и жевательные мышцы. Примечательно, что скорость прогрессирования болезни и выраженность ограничения движений не зависели от возраста дебюта болезни [7].
Гистопатология пораженных тканей при ПОФ хорошо описана в литературе [8–12]. Исследование тканей ранних проявлений ПОФ говорит об интенсивной мононуклеарной и периваскулярной инфильтрации моноцитов, макрофагов, мастоцитов, T- и B-клеток. Их точная роль в эволюции обострений ПОФ неизвестна, хотя установлено, что любое локальное воспаление независимо от причины его возникновения может спровоцировать активизацию болезни.
Последующая миграция одноядерных воспалительных клеток в пораженную мышечную ткань предшествует масштабной гибели скелетных мышц – процессу, после которого происходит формирование гетеротопического эндохондрального зачатка. За скоротечной воспалительной стадией следует период интенсивной фибробластной пролиферации, сопровождаемой мощным ангиогенезом. Фибробластическое поражение на раннем этапе в отношении гистологии неотличимо от агрессивного ювенильного фиброматоза, но по мере созревания ткани фиброзная ткань проходит через фазу бессосудистого уплотнения до состояния хряща, а затем наступает этап реваскуляризации с остеогенезом и характерным процессом эндохондральной оссификации.
Все стадии гистологического развития активных патологий указывают на то, что различные участки пораженного участка ПОФ проходят полный процесс эндохондральной оссификации с разной скоростью. Примечательно, что образующаяся в результате новая гетеротопическая кость гистологически выглядит как нормальная сформировавшаяся кость и часто содержит включения костного мозга. Мастоциты обнаруживаются на всех этапах формирования тканей при поражении ПОФ, и их количество значительно выше, чем в обычных скелетных мышцах или мышечных тканях больного вне зоны поражения ПОФ. При поражениях ПОФ на этапе фибробластной пролиферации количество мастоцитов многократно превышает показатель, свойственный любой другой форме воспалительной миопатии [13].
При том, что процесс гетеротопного костеобразования в некоторых отношениях схож с формированием костей скелета при эмбриональном развитии или постнатальном заживлении переломов, важнейшим отличием является отсутствие воспалительного компонента при первичном скелетообразовании. Впрочем, возможно, эти различия вполне условны и объясняются недостаточным объемом накопленных данных о морфологии ПОФ на различных стадиях болезни и малой перспективностью их дополнения, поскольку проведение биопсии пораженных (и даже клинически непораженных) тканей, проведение любого, даже малотравматичного хирургического вмешательства сопряжено с исключительно высоким риском образования новых очагов и быстрого прогрессирования болезни. Хотя процесс гетеротропического костеобразования в некоторых отношениях схож с формированием костей скелета при эмбриональном развитии и послеродовом заживлении переломов, важнейшим отличием является отсутствие воспалительного компонента при первичном скелетообразовании.
Генетика ПОФ и семейные исследования. Семейная ПОФ – чрезвычайно редкое заболевание в популяции и имеет аутосомно-доминантный тип наследования с полной пенетрантностью [14]. Хотя большинство зарегистрированных случаев заболевания являются спорадическими (единичными в семье), в литературе описаны семьи, в которых поражены родитель – потомок/потомки: братья, сестры, в т. ч. близнецы. Представляет интерес наблюдение J.M. Connor et al. [15], описавших семью, где ПОФ была диагностирована в 3-х поколениях у 5 лиц. В 1-м поколении пораженным был мужчина, который имел 2-х больных дочерей и 2-х больных внучек. Примечательна выраженная фенотипическая изменчивость тяжести течения заболевания в этой семье. Так, мужчина имел асимптоматическое течение до момента травмы, после которой у него развилась тугоподвижность шеи, спины, конечностей и нижней челюсти. Однако в 70 лет он передвигался, используя трость, и умер в возрасте 72 лет от инфаркта миокарда. Диагноз ПОФ ему был поставлен на основании наличия врожденной деформации большого пальца стопы и рентгенологического обследования, выявившего генерализованный анкилоз позвоночника, множественные зоны оссификации мягких тканей. Одна из его дочерей также не имела признаков заболевания до 22 лет, когда после экстракции зуба с последующим протезированием у нее развилась тугоподвижность челюсти. Ее сестра в возрасте 15 лет отметила спонтанную припухлость левой голени, когда на основании биопсии была диагностирована фибросаркома. Однако дальнейшее течение болезни (характерные болезненные припухлости и прогрессирующее ограничение объема движений) свидетельствовало в пользу ПОФ. Летальный исход наступил в 28 лет от пневмонии в состоянии полной обездвиженности. Также имели ПОФ 2 внучки больного от старшей дочери.
F.S. Kaplan et al. [16] представили данные о передаче ПОФ от пораженного отца 2-м дочерям и сыну. Так, 27-летний мужчина (от брака клинически здоровых родителей, 3-й сын в семье, 2 брата здоровы), имеющий характерную микродактилию большого пальца, заболел в 2 года после травмы. Клинически наблюдалось появление болезненных узлов в области шеи с последующей генерализацией процесса и эктопической оссификацией, прогрессированием болезни и формированием значительного двигательного дефицита. Рентгенологически определялись укорочение и расширение шеек бедренных костей. У 3-х его потомков также присутствовала врожденная микродактилия больших пальцев с дальнейшим развитием типичной картины ПОФ, при этом у одного из его сыновей первые оссификаты четырехглавой мышцы бедра появилась в 2-месячном возрасте после в/м инъекции, у 2-х других детей – в 3-летнем возрасте.
Как свидетельствуют и другие литературные наблюдения [17], авторами часто описывается фенотипическая гетерогенность ПОФ: характер послеродовой гетеротопической оссификации отличается в зависимости от анамнеза, случаев травматизации мягких тканей и заболеваемости вирусными болезнями. По мнению N. Hebela et al. [18], формирование фенотипа заболевания в период пренатального развития определяется генетическими факторами, в то время как сопутствующие (средовые) факторы влияют на послеродовой ход гетеротопической оссификации.
Молекулярная генетика. Е.М. Shore et al. [19] провели полногеномный анализ сцепления на материале 5 семей с ПОФ и выявили сцепление заболевания с регионом 23–24 хромосомы 2 (2q 23-q24), в котором картируется ген ACVR1 (Activine A Rreceptor, type I) (альтернативное название ACK2 – Activine Receptor-like Kinase 2). Секвенирование всех белок-кодирующих экзонов и экзон-интронных областей позволило ученым выявить гетерозиготную мутацию R(206)H (замена аминокислоты аргинин на гистидин в позиции 206) у всех пораженных ПОФ членов 5 семей, а также 32 больных со спорадическим ПОФ. Исследование молекулярно-генетических основ ПОФ в различных популяциях позволило установить, что заболевание тесно сцеплено главным образом с гетерозиготной мутацией de novo R(206)H при спорадической ПОФ [20–22]. Также обнаружена гетерозиготная миссенс-мутация de novo G(356)D в этом же гене [23].
Исследования показали, что во всех случаях спорадических и семейно-наследственных заболеваний у пациентов с классической клинической картиной ПОФ выявлена мутация R(206)H в гене ACVR1/ALK [24–26]. Данный ген кодирует рецептор ACVR1, относящийся к семейству BMP (bone morphogenetic protein)-рецепторов. BMP – регуляторный белок, участвующий в процессе эмбрионального формирования и постнатального восстановления костей скелета (индуцирует развитие мезодермы, формирование костной ткани, зубов, костеобразование и репарацию переломов) [27]. Преобразование сигналов ВМР происходит через рецепторы типа 1: BMPR1А, BMPR1B и ACVR1/ALK2 [28].
A.B. Shafrits et al. [29] обнаружили повышенные уровни белка BMP4 и его экспрессии (мРНК) в лимфобластоидных клетках у 26 из 32 больных с ПОФ и у 1 из 12 здоровых лиц (р<0,001). Авторами выявлены эти изменения также у больного мужчины и 3-х его детей, страдающих ПОФ, в то время как среди его здоровых потомков такой закономерности не выявлено.
Кроме того, естественным эндогенным фактором, ингибирующим экспрессию гена BMP, является Noggin-протеин, синтез которого кодирует ген NOG. Мутации в гене NOG были выявлены у больных с проксимальным симфалангизмом, фенотипически схожим с дефектами пальцев при ПОФ. Исходя из этого G. Lucotte et al. [30], исследуя последовательность пар оснований (секвенирование) гена NOG, выявили у больного ПОФ наличие гетерозиготной делеции (утраты) 42 пар оснований в белок-кодирующем сегменте этого гена. Предположено, что такой ген может кодировать синтез укороченного белка, который будет неэффективно ингибировать экспрессию ВМР, а он в свою очередь будет суперэкспрессирован.
Представляет интерес гипотеза механизма обострения на фоне травматизации, основанная на результатах исследования мастоцитов. Так, травма у больного ПОФ провоцирует миграцию макрофагов, мастоцитов и лимфоцитов в нормальные скелетные мышцы. Медиаторы, выделяемые мастоцитами, в свою очередь стимулируют развитие воспалительного отека, фиброза и ангиогенеза, который усиливается, находясь в авангарде патологических изменений, связанных с ПОФ. Реактивно активизированные фибробласты мышечной ткани продуцируют протеины, вызывая дальнейшее распространение мастоцитов и самоподдерживающееся нарастание процесса, известного под названием обострения ПОФ. В норме трансформирующий фактор роста-β, выделяемый мастоцитами и клетками-предшественниками соединительной ткани, ограничивает процесс привлечения и миграции иммунных клеток, а значит, размер и степень распространения поражения тканей, в то время как эндогенная сверхактивность ACVR1/ALK2 направляет этот процесс в сторону оссификации [31].
Недавние исследования больных (n=254) с различными поражениями мягких тканей [32] позволяют обсуждать роль DMP-1 (Dentine matrix protein 1) как маркера остеогенных доброкачественных и злокачественных опухолей и опухолеподобных поражений, когда наблюдаются формирование костной ткани и минерализация матрицы. Так, DMP-1 определялся практически во всех случаях оссификаций, фиброзно-костных опухолей, в плотной коллагеновой соединительной ткани при ее патологической минерализации, а также в случаях кальциноза мягких тканей при дерматомиозите, кальцинозе сухожилий, в областях фокусной оссификации и кальцификации при синовиальной саркоме и других опухолях мягких тканей. Авторы отмечают целесообразность изучения DMP-1 как с дифференциально-диагностической целью между внескелетными остеосаркомами и другими видами сарком, так и при гетеротопической оссификации/минерализации мягких тканей, в т. ч. при ПОФ.
ПОФ дебютирует, как правило, в детском возрасте и носит прогрессирующий характер. Нередко заболевание манифестирует повышением температуры тела и появлением болезненных уплотнений и припухлости в мышцах на спине, области шеи с последующими их кальцификацией и оссификацией, ограничивающими движения, вплоть до обездвиженности и превращения людей в «живые статуи». Гетеротопическая оссификация происходит в направлении от оси к периферии: от головы к конечностям, от проксимальных отделов к дистальным. К зонам ранней локализации гетеротопической оссификации относятся: шея, позвоночник, плечевой пояс и жевательные мышцы. Образование этих инфильтратов может провоцироваться ушибами, оперативными вмешательствами [33], в/м и подкожными инъекциями, в т. ч. при выполнении плановых вакцинаций, однако патологические изменения могут возникать и без видимых причин [34]. В то же время обострение заболевания может быть спровоцировано не только травмами, но и гриппоподобными вирусными заболеваниями [35] либо возникать спонтанно.
Прогрессирующее течение ПОФ с увеличением зон оссификации ведет к тяжелым осложнениям, включая кривошею с вовлечением musculus sternoclaidomastoideus, деформации верхней половины грудной клетки, сколиоз и тугоподвижность суставов, нарушение жевания и даже резкое ограничение открытия рта, серьезно затрудняющее процесс приема пищи. Тяжелое поражение жевательной мускулатуры часто провоцируется стоматологическими манипуляциями, особенно выполнением инъекций анестетиков.
Продолжительность жизни больных различна. Значительная часть больных умирает до 20 лет. В качестве наиболее частой причины летального исхода выступает легочная инфекция на фоне гиповентиляции вследствие поражения межреберных мышц. Описаны случаи смертельного исхода вследствие алиментарного истощения, обусловленного окостенением жевательной и глотательной мускулатуры.
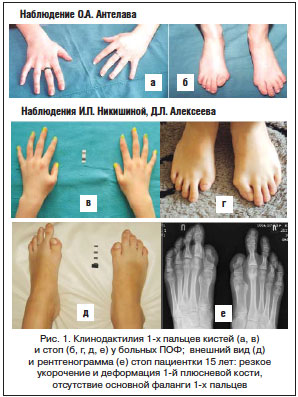
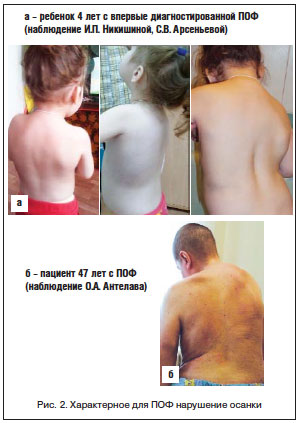
Диагностика ПОФ в дебюте болезни, как правило, бывает ошибочна. Наиболее часто заболевание диагностируется только после развития эктопической оссификации, которая наступает в возрастном диапазоне от 0 до 14 лет (в среднем после 2,7 года от начала заболевания). Однако при достаточной осведомленности врача диагностика ПОФ не вызывает трудностей. Уже в первые дни жизни обращают на себя внимание врожденные скелетные аномалии: микро- или клинодактилия первых пальцев кистей и/или стоп (рис. 1 а–е), адактилия первых пальцев стоп. В дальнейшем формируется характерная поза больного, обусловленная формированием кривошеи и оссификацией скелета (рис. 2 а, б). Выявляются также анкилоз первых межфаланговых суставов (95%), реже – глухота, облысение волосистой части головы, слабовыраженная задержка психического развития [36]. Анамнестические данные (болезненные уплотнения с характерной локализацией в дебюте болезни) также способствуют ранней правильной диагностике. Рентгенографическая картина, описанная F.S. Kaplan (рис. 3) [37], включает характерные для данного заболевания изменения: сращение (monophalangism) первых фаланг стоп и их вальгусную деформацию (рис. 3 а, б), синостоз шейных позвонков (рис. 3 в, рис. 4 а, б), укорочение и расширение шеек плечевых и бедренных костей (рис. 3 г), остеохондромы проксимальных отделов большеберцовых костей (рис. 3 д), дисплазию метафизов, синостозы реберно-позвоночных сочленений, экзостозы позвоночника и метафизов длинных костей. Результаты биохимических исследований минерального обмена костной ткани обычно находятся в пределах нормы, хотя активность щелочной фосфатазы и СОЭ могут быть несколько повышены, преимущественно в периоды обострения болезни. Уровень повышения основного фактора роста фибробласта в моче может повышаться в период обострений, совпадающих по времени с костно-ангиогенной фазой раннего этапа фибропролиферации.
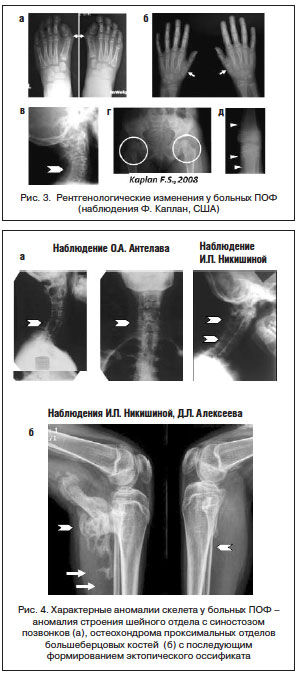
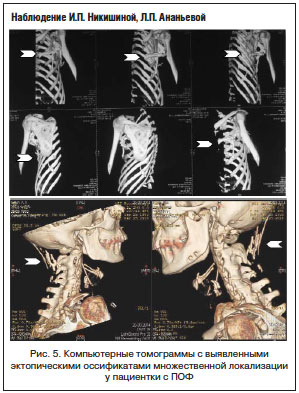
В круг дифференциального диагноза обычно включаются как доброкачественные, так и злокачественные новообразования костей и мягких тканей, чаше всего – фибросаркома, имеющая некоторые сходные морфологические черты, приводящие в заблуждение: разрастание молодой, богатой клеточными элементами соединительной ткани с единичными мышечными волокнами, наличие крупных, уродливых фибробластов с гиперхромными ядрами, зернистой цитоплазмой. При ПОФ обнаруживаются частично обызвествленные костные балки, обычно расположенные на периферии узла и имеющие остеоидное строение. Случаи подобных диагностических ошибок описаны в литературе и встречались в нашей практике. Так, с 2003 г. специалистами детского отделения Института ревматологии наблюдается пациентка, которой диагноз ПОФ был установлен в возрасте 9 лет после длительного периода безуспешного лечения у онкологов с ошибочно диагностированной рабдомиосаркомой, по поводу чего девочка получила 11 сеансов химиотерапии и 7 сеансов лучевой терапии. Гистологические препараты были пересмотрены нашими морфологами, и с учетом анализа анамнеза и клинической картины диагноз был изменен на ПОФ. В последующем этот диагноз был подтвержден и результатами молекулярно-генетического исследования. Примечательно, что на момент установки диагноза и в последующие 8 лет лучевыми методами исследования не выявлялось наличие эктопических оссификатов, несмотря на наличие типичных клинических проявлений «деревянистой» плотности уплотнений подкожных тканей шеи, плечевого пояса и спины. Первые очаги оссификатов были выявлены в возрасте 17–18 лет, что наглядно демонстрирует рисунок 5.
Убедительных данных об эффективности какой-либо терапии, в т. ч. глюкокортикоидами, применение которых обсуждается в период обострения [38], не существует. В прошлом широко использовалась этилендиаминтетрауксусная кислота (ЭДТА) [39], образующая комплексные соединения с катионами и ионами кальция, однако ее применение при ПОФ не позволяет достичь желаемых результатов.
Применение бисфосфонатов рассматривается как возможное средство лечения ПОФ и других заболеваний с сопутствующей гетеротопической оссификацией [40]. Первый из них – этидронат, который при введении в больших дозах оказывает мощное ингибирующее действие на процесс минерализации вновь образующихся хрящей и протеинов. Однако, к сожалению, высокие дозы введенного в/в препарата приводили к остеомаляции не только гетеротопических оссификаций, но и всей скелетной структуры организма, что ограничило возможность использования подобной схемы лечения. Применение бисфосфонатов перорально остается распространенным в терапевтической практике, описано применение памидроновой, золедроновой и алендроновой кислот.
Обсуждается применение миорелаксантов для уменьшения вторичного спастического мышечного напряжения и купирования боли [41] и кроормолина – стабилизатора мастоцитов, роль которых в патогенезе болезни обсуждается [42].
Как известно, провоспалительные простагландины являются мощными молекулярными ко-стимуляторами оссификации, что позволяет рассматривать роль селективных ингибиторов циклооксигеназы-2 в симптоматическом лечении ПОФ [43, 44]. Так, снижение уровня провоспалительных простагландинов у животных резко повышает порог запуска процесса гетеротопической оссификации, усложняя условия ее формирования, а их уровень в моче больных ПОФ резко повышен, особенно в периоды обострения болезни [45]. Следовательно, снижение их уровня позволит повысить порог формирования гетеротопической оссификации даже в присутствии хаотично активных ACVR1/ALK2 [46].
Генетические исследования стали важной вехой на пути понимания механизма болезни и поиска терапевтической мишени в сигнальной системе ВМР, разработки ее ингибиторов, функционирующих через посредство ACVR1/ALK2. Так, проводятся экспериментальные работы по изучению блокаторов мутации в гене ACVR1/ALK2 и эффективности ретиноевой кислоты при гетеротопической оссификации (как генетически обусловленной, так и посттравматической) [47].
Несмотря на довольно пессимистические прогнозы, некоторые пациенты при самоотверженной поддержке и уходе близких доживают до весьма зрелого возраста, о чем свидетельствует следующее клиническое наблюдение.
Клинический пример
Под нашим наблюдением находится больной Т., 1967 г. р.
Диагноз: ПОФ, генерализованная гетеротопическая оссификация. Артериальная гипертензия. Инвалид 1-й группы с 1999 г.
Отец, мать, родная сестра и племянник – клинически (фенотипически) здоровы. Члены семьи неоднократно переносили оперативные вмешательства различного объема и степени тяжести, отец занимался спортом, при этом клинических признаков ПОФ не развивалось.
При осмотре обращало на себя внимание некоторое укорочение первых пальцев стоп у отца (более выраженных, чем непосредственно у больного), однако, со слов родственников, все члены семьи со стороны отца также здоровы.
Диагноз ПОФ был поставлен в дошкольном возрасте на основании клинической картины и анамнестических данных больного.
Течение болезни – классическое для ПОФ: очаги инфильтрации в области шеи, на спине с последующей оссификацией. В детском возрасте больной посещал школу, стараясь «превозмочь» болезнь, активно занимался спортом (!). Тем не менее болезнь неуклонно прогрессировала, зоны оссификации увеличивались, инвалидизация возрастала, самообслуживание становилось все более затруднительным.
С 18-летнего возраста и в течение последующих 5 лет – ежегодные госпитализации по месту жительства, проводилась терапия ЭДТА (в/в, капельно) № 15 – без видимого эффекта; получал этидроновую кислоту, с 2005 г. – алендроновую кислоту.
В настоящее время (2015 г.) пациенту 47 лет, он полностью обездвижен и нуждается в постоянном уходе. Положение вынужденное, неестественное: кривошея, контрактуры локтевых, тазобедренных и коленных суставов, фиксация их в состоянии неполного разгибания. Некоторую подвижность сохраняют только пальцы кистей (рис. 6), благодаря чему больной может читать и работать за компьютером в положении стоя. Для подъема пациента с кровати необходимо участие нескольких человек, поскольку из-за генерализованной оссифицикации активное движение практически невозможно (рис. 7).
Питание затруднено – в связи с оссификацией жевательных мышц ротовая щель открывается не более чем на 2–3 мм, поэтому кормление проводится при помощи специальных приспособлений, однако аппетит не нарушен.
Сегодня прогрессирование болезни продолжается, происходят обострения как в весенне-осенний период, так и на фоне простудных заболеваний – появляются болезненные припухлости и гиперемия различных локализаций, сопровождающиеся субфебрилитетом.
Пациент обладает высоким интеллектом, в 21 год окончил авиационный колледж (дальнейшая учеба была невозможна из-за нарастания инвалидизации), принимал участие в образовании племянника и помогал в подготовке его дипломной работы в университете.
Рентгенологическое обследование больного уже в 2005 г. было невозможно из-за вынужденного фиксированного положения туловища. В анализах отклонений от нормы не отмечается: гемоглобин – 141 г/л, лейкоциты – 8,2х109, эритроциты – 5,4х1012, СОЭ – 5 мм/ч, глюкоза – 5,1, протромбиновый индекс – 100%. ЭКГ: ритм синусовый, горизонтальное положение электрической оси сердца, обменные изменения задней стенки левого желудочка.
В настоящее время получает гипотензивную терапию: гидрохлоротиазид+лозартан 50 мг/сут, с 2005 г. – алендроновую кислоту 70 мг/нед., в период обострения – нестероидные противовоспалительные препараты и троксерутин (местно).
Таким образом, ПОФ остается одним из тяжелых, неуклонно прогрессирующих заболеваний, требующих как поиска новых терапевтических возможностей, так и осведомленности (ранняя диагностика) и правильной врачебной тактики во избежание попыток хирургической коррекции или взятия биоптатов и предотвращения ятрогенных поражений, а также оптимизации оставшихся функциональных возможностей больного.
Литература
- Wortman R.L. Inflammatory Diseases on Mascle // Textbook of Reumatology. Fourth edition. 1993. Vol. II. P. 1170.
- Fibrodisplasia (myositis) ossificans progressiva. Primer on metabolic bone diseuses and disoders of mineral metabolism. Fierst edition. Editor: M.J. Favus. 1990. Р. 267–269.
- Wheeless C.R. Myositis Ossificans (Sterner’s Tumor). Wheeless’ Text book of Ortthopaedics. http://www.nedug.ru/lib/traum/wheeless/93-94.
4. Hegyi L., Gannon F.H., Glaser D.L., Shore E.M., Kaplan F.S., Shanahan C.M. Stromal cells of fibrodysplasia ossificans progressiva lesions express smooth muscle lineage markers and the osteogenic transcription factor Runx2/Cbfa-1: clues to a vascular origin of heterotopic ossification? // Pathol. 2003. Vol 201. N 1. Р. 141–148. - Kaplan F.S., Le Merrer M., Glaser D.L., Pignolo R.J., Goldsby R., Kitterman J.A., Groppe J., Shore E.M. Fibrodysplasia ossificans progressiva. Best Practice & Research // Clinical Rheumatology. 2008. Vol. 22. Р. 191–205.
- Крисюк А.П., Городняя В.Н., Салдимирова Л.Я. Способ лечения прогрессирующегооссифицирующегомиозита // Киевский институт ортопедии. 1983. 9 с.
- Smith R., Athanasou N.A., Vipond S.E. Fibrodysplasia (myositis) ossificans progressiva: clinicopathological features and natural history // Quart. J. Med. 1996. Vol. 89. P. 445–456.
- Kaplan F.S., Tabas J., Gannon F.H., Finkel G., Hahn G.V., Zasloff M.A. The histopathology of fibrodysplasia ossificans progressiva: an endochondral process // J Bone Joint Surg Am. 1993. Vol. 75A. P. 220–230.
9. Gannon F.H., Valentine B.A., Shore E.M., Zasloff M.A., Kaplan F.S. Acute lymphocytic infiltration in an extremely early lesion of fibrodysplasia ossificans progressiva // Clin Orthop. 1998. Vol. 346. P. 19–25. - Hegyi L., Gannon F.H., Glaser D.L., Shore E.M., Kaplan, F.S., Shanahan C.M. Stromal cells of fibrodysplasia ossificans progressiva lesions express smooth muscle lineage markers and the osteogenic transcription factor Runx2/Cbfa-1: Clues to a vascular origin of heterotopic ossification // J Pathol. 2003. Vol. 201. P. 141–148.
- Pignolo R.J., Suda R.K., Kaplan F.S. The fibrodysplasia ossificans progressiva lesion // Clin Rev Bone Miner Metab. 2005. Vol. 3. P. 195–200.
- Антелава О.А., Лобжанидзе Т.Б., Никишина И.П., Федоров Е.С., Бельская Е.А., Хитров А.Н. Прогрессирующаяоссифицирующаяфибродисплазия // РМЖ. 2005. № 8. C. 560–564.
- Gannon F.H., Valentine B.A., Shore E.M., Zasloff M.A., Kaplan F.S. Acute lymphocytic infiltration in an extremely early lesion of fibrodysplasia ossificans progressiva // Clin Orthop. 1998. Vol. 346. P. 19–25.
- OMIM#135100 – Online Mendelian Inheritance in Man.
- Connor J.M., Skirton H., Lunt P.W. A three generation family with fibrodysplasia ossificans progressiva // J. Med. Genet. 1993. Vol. 30. P. 687–689.
- Kaplan F.S., McCluskey W., Hahn G. et al. Genetic transmission of fibrodysplasia ossificans progressiva: report of a family // J. Bone Joint Surg. 1993. Vol. 75A. P. 1214–1220.
- Janoff H.B., Muenke M., Johnson L.O., Rosenberg A., Shore E.M., Okereke E., Zasloff M., Kaplan F.S. Fibrodysplasia ossificans progressiva in two half-sisters: evidence for maternal mosaicism // Am J Med Genetics. 1996. Vol. 61. P. 320–324.
- Hebela N., Shore E.M., Kaplan F.S. Three pairs of monozygotic twins with fibrodysplasia ossificans progressiva: the role of environment in the progression of heterotopic ossification // Clin Rev Bone Miner Metab. 2005. Vol. 3. P. 205–208.
- Shore E.M., Xu M., Feldman G.J., Fenstermacher D.A., Cho T.-J., Choi I.H., Connor J.M., Delai P., Glaser D.L., LeMerrer M., Morhart R., Rogers J.G., Smith R., Triffitt J.T., Urtizberea J.A., Zasloff M., Brown M.A., Kaplan F.S. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva // Nature Genet. 2006. Vol. 38. P. 525–527.
- Lin G.-T., Chang H.-W., Liu C.-S., Huang P.-J., Wang H.-C., Cheng Y.-M. De novo 617G-A nucleotide mutation in the ACVR1 gene in a Taiwanese patient with fibrodysplasia ossificans progressiva // J. Hum. Genet. 2006. Vol. 51. P. 1083–1086.
- Bocciardi R., Bordo D., Di Duca M., Di Rocco M., Ravazzolo R. Mutational analysis of the ACVR1 gene in Italian patients affected with fibrodysplasia ossificans progressiva: confirmations and advancements // Europ. J. Hum. Genet. 2009. Vol. 17. P. 311–318.
- Nakajima M., Haga N., Takikawa K., Manabe N., Nishimura G., Ikegawa S. The ACVR1 617G-A mutation is also recurrent in three Japanese patients with fibrodysplasia ossificans progressiva // J. Hum. Genet. 2007. Vol. 52. P. 473–475.
- Furuya H., Ikezoe K., Wang L., Ohyagi Y., Motomura K., Fujii N., Kira J., Fukumaki Y. A unique case of fibrodysplasia ossificans progressiva with an ACVR1 mutation, G356D, other than the common mutation (R206H) // Am. J. Med. Genet. 2008. Vol. 146A. P. 459–463.
- Nakajima M., Haga N., Takikawa K. еt al. The ACVR1 617G>A mutation is also recurrent in three Japanese patients with fibrodysplasia ossificans progressiva // J. Hum. Genet. 2007. Vol. 52 (5). P. 473–475.
25. Koster B., Pauli R.M., Reardon W. еt al. Classic and atypical fibrodysplasia ossificans progressiva (FOP) phenotypes are caused by mutations in the bone morphogenetic protein (BMP) type I receptor ACVR1 // Hum. Mutat. 2009. Vol. 303. P. 79–90. - Deirmengian J.K., Hebela N.M., O’Connell M., Glaser D.L., Shore E.M. et al. Proximal Tibial Osteochondromas in Patients with Fibrodysplasia Ossificans Progressiva // J Bone Joint Surg Am. 2008. Vol. 90 (2). P. 366–374.
- Groppe J.C., Shore E.M., Kaplan F.S. Functional Modeling of the ACVR1 (R206H) mutation in FOP // Clin. Orthop. Relat. Res. 2007. Vol. 462. P. 87–92.
- Rigueur D., Brugger S., Anbarchian T., Kim J.K., Lee Y.J., Lyons K.J. The type I BMP receptor ACVR1/ALK2 is required for chondrogenesis during development // Bone Miner Res. 2014. Nov 21.
- Shafritz A.B., Shore E.M., Gannon F.H. et al. Overexpression of an osteogenic morphogen in fibrodysplasia ossificans progressiva // New Eng. J. Med. 1996. Vol. 335. P. 555–561.
- Lucotte G., Semonin O., Lutz P. A de novo heterozygous deletion of 42 base-pairs in the noggin gene of a fibrodysplasia ossificans progressiva patient. (Letter) // Clin. Genet. 1999. Vol. 56. P. 469–470.
- Gannon F.H., Glaser D., Caron R., Thompson L.D.R., Shore E.M., Kaplan F.S. Mast cell involvement in fibrodysplasia ossificans progressiva // Hum Pathol. 2001. Vol. 32. P. 842–848.
- Inagaki Y.L, Kashima T.G., Hookway E.S., Tanaka Y., Hassan A.B., Oppermann U., Athanasou N.A. Dentine matrix protein 1 (DMP-1) is a marker of bone formation and mineralisation in soft tissue tumours // Virchows Arch. 2015. Jan 29.
- Коваленко-Клычкова Н.А., Клычкова И.Ю., Кенис В.М., Мельченко Е.В. Прогрессирующая оссифицирующая фибродисплазия у детей (обзор литературы и анализ 5 клинических случаев) // Травматология и ортопедия России. 2014. № 1. С. 102–109.
- Зацепин С.Т. Костная патология взрослых. М.: Медицина, 2001. 639 с.
- Scarlett R.F., Rocke D.M., Kantanie S. еt al. Influenzalike viral illnesses and flare-ups of fibrodysplasia ossificans progressiva // Clin. Orthop. Relat. Res. 2004. Vol. 423. P. 275–279.
- Рябова Т.В., Геппе Н.А., Михалева Г.В., Сермягина И.Г. Прогрессирующая оссифицирующая фибродисплазия у детей // Вопросы практической педиатрии. 2011. № 6(2). С. 99–106
- Kaplan F.S., Xu M., Glaser D.L., Collins F., Connor M., Kitterman J., Sillence D., Zackai E., Ravitsky V., Zasloff M., Ganguly A., Shore E.M. Early diagnosis of fibrodysplasia ossificans progressiva // Pediatrics. 2008. Vol. 121 (5). P. 1295–300.
- Rhen T., Cidlowski J.A. Anti-inflammatory action of glucocorticoids – new mechanisms for old drugs // N Engl J Med. 2005. Vol. 353. P. 1711–1723.
- Динатриевая соль этилендиаминтетрауксусной кислоты. http:/www.pharmasvit.com /v3/ Spravochniki/lecarstvo1293.html.
40. Orcel P., Beaudreuil J. Bisphosphonates in bone disease other than osteoporosis // Joint Bone Spine. 2002. Vol. 69. P. 19–27. - Glaser D.L., Kaplan F.S. Treatment considerations for the management of fibrodysplasia ossificans progressiva // Clin Rev Bone Miner Metab. 2005. Vol. 3. P. 243–250.
- Gannon F.H., Glaser D., Caron R., Thompson L.D.R., Shore E.M., Kaplan F.S. Mast cell involvement in fibrodysplasia ossificans progressiva // Hum Pathol. 2001. Vol. 32. P. 842–848.
- Weinreb M., Suponitsky I., Keila S. Systemic administration of an anabolic dose of PGE2 in young rats increases the osteogenic capacity of bone marrow // Bone. 1997. Vol. 20. P. 521–526.
- Jones M.K., Wang H., Peskar B.M., Levin E., Itani R.M., Sarfeh I.J., Tarnawski A.S. Inhibition of angiogenesis by nonsteroidal anti-inflammatory drugs: insight into mechanisms and implications for cancer growth and ulcer healing // Nature Med. 1999. Vol. 5. P. 1418–1423.
- Levitz C.L., Cohen R.B., Zasloff M.A., Kaplan F.S. The role of prostaglandins in bone formation // Abstracts from The First International Symposium on Fibrodysplasia Ossificans Progressiva. September 25–26, 1991, Philadelphia, Pennsylvania // Calcif Tissue Int. 1992. Vol. 50. P. 385–388.
- DiCesare P.E., Nimni M.E., Pen L., Yazdi M., Cheung D.T. Effects of Indomethacin on demineralized bone-induced heterotopic ossification in the rat // J Orthop Res. 1991. Vol. 9. P. 855–861.
- Shimono K., Tung W.E., Macolino C., Chi A.H., Didizian J.H., Mundy C. et al. Potent inhibition of heterotopic ossification by nuclear retinoic acid receptor-gamma agonists // Nat. Med. 2011. Vol. 17. P. 454–460.



Комментировать