Синдром Ниймеген: общая характеристика. Описание случаев из медицинской практики.
О.Л. Цимбаліста, Н.М. Фоменко, Г.С. Бенько, В.Б. Дехтяр, О.В. Пастух, О.С. Бобрикович Івано-Франківський державний медичний університет,Івано-Франківська обласна дитяча клінічна лікарня
Резюме. Синдром Ниймеген принадлежит к тяжелым комбинированным иммунодефицитным заболеваниям с аутосомнорецессивным типом наследования. В основе заболевания лежит
мутациягенаNBS1, расположенногоонсаом8ех.ром Наиболее характерными фенотипическими признаками заболевания являются микроцефалия, «птицеобразное» лицо, задержка физического развития и полового созревания, нарушения пигментации кожи. Клиническая манифестация заболевания проявляется рецидивирующими вирусно—бактериальными инфекциями, злокачественными новообразованиями, регрессом интеллекта
Вступ
Поширеність імунодефіцитних станів (ІДС), проявом яких є атиповий ускладнений перебіг багатьох захворювань, сприяє пошуку нових діагностичних програм з метою раннього виявлення ІДС [1]. Досягнення сучасної клінічної генетики та імунології дозволяють у багатьох випадках встановити нозологічну форму спадково зумовленого імунодефіцитного захворювання, скласти програму лікування та реабілітації хворої дитини та запобігти повторному народженню в даній сім‘ї дітей з аналогічним захворюванням.
Найуспішнішою є діагностика імунодефіцитних захворювань, які мають характерні клініко-фенотипові маркери. До таких хворювань
насамперед належить синдром Ніймеген.
Синдром Ніймеген є рідкісним аутосомно—рецесивним захворюванням, що виникає в результаті порушення репарації ДНК. Захворювання було вперше описане в 1981 р. Найчастіше ця патологія зустрічається в країнах Східної Європи (Польща, Чехія, Україна). В основі цього ІДС лежить мутація гена NBS1, розташованого на 8 хромосомі (8q21). Захворювання має аутосомно—рецесивний тип успадкування. Ген NBS1 кодує нібрин — білок, що бере участь у процесах репарації ДНК, контролює процеси мітозу. Порушення репарації веде до нагромадження ушкоджень ДНК і до розвитку імунодефіциту, злоякісних новоутворень. Відмічається підвищенаислхьність до ушкоджень 7 і 14 хромосом, що несуть гени Т—клітинного рецептора, важких ланцюгів молекул імуноглобулінів.
Більше ніж у 90% випадків у хворих в обох локусах відмічається однакова мутація гена NBS1, (657del5, або «Слов‘янська мутація»). Описано 7 інших мутацій цього гена. У всіх випадках пацієнти були гетерозиготами.
В клінічній картині для всіх дітей характерні: мікроцефалія, яка виявляється з моменту народження і прогресує з віком; інші ураження кавернозні гемангіоми; у дітей раннього віку волосся рідке, але з віком ріст волосся нормалізується, характерною є передчасна сивина; аномалії розвитку інших органів і систем. Проявами імунодефіциту є рецидивні інфекції дихальних шляхів зі схильністю до хронізації і формування бронхоектазів. Часто спостерігаються рецидивні інфекції нирок, сечовидільних шляхів, кишкові інфекції. Опортуністичні інфекції зустрічаються рідко.
Найчастішою причиною смерті цих пацієнтів є злоякісні новоутворення, частота яких значно перевищує таку серед загальної популяції дітей. Більшість злоякісних новоутворень розвиваються у віці до 9 років. За частотою: лімфоми, гостра лімфобластна лейкемія, лімфогранулематоз.
Діагностичними критеріями є: типова клініка, генетичне обстеження пацієнтів на предмет виявлення «слов‘янської мутації» та імунологічні дослідження (лімфопенія, переважно за рахунок CD4+-лімфоцитів, інверсії співвідношення CD4+/CD8+, підвищення вмісту NKлімфоцитів, зниження утворення імуноглобулінів, що варіює від ізольованого селективного дефіциту IgA, дефіциту субкласів IgG до гіпогаммаглобулінемії, рівень IgM нормальний чи навіть підвищений. Лікування: розглядається питання трансплантації стовбурових клітин, замісна терапія внутрішньовенним імуноглобуліном [3].
Матеріал і методи дослідження
Метод генеалогічного аналізу, оцінка фенотипу, загальноклінічне, імунологічне обстеженя (гуморальної та клітинної ланок імунітету), спеціальне генетичне обстеження на найбільш поширені мутації гена данного захворювання NBS1 на базі Львівського інституту спадкової патології АМН України, медична документація (медико—генетичні карти, історії стаціонарних хворих, історії розвитку дітей).
Результати досліджень та їх обговорення
В популяції Прикарпаття за останні 6 років синдром Ніймеген діагностовано у чотирьох дітей, з них двоє дітей з однієї сім’ї. У двох дітей діагноз синдрому вста новлено на підставі харак- терного фенотипу (мік роцефалія, нанізм, харак- терні лицьові дизмор фії, відносно легке відста- вання у психоінтелекту альному розвитку, прояви імунодефіциту та наявн ість злоякісного захворю- вання лімфоретикуляр ної системи). З них одна дитина померла у віці трьох років від лімфогра нулематозу; друга — у віці 12 років від лімфобластної лейкемії. Онкоге матологічні захворювання були встановлені кліні чно і верифіковані за допомогою патологоанатомічного дослідження. Хоча спеціального генетичного обстеження не виконано (на той час не проводилося), діагноз синдрому Ніймеген не в икликає жодного сумніву
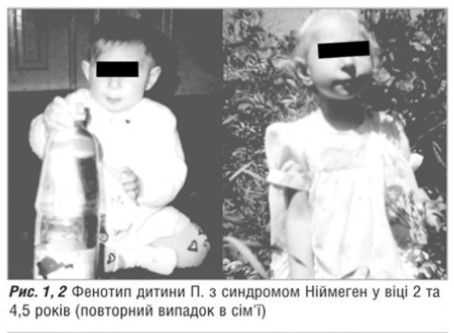
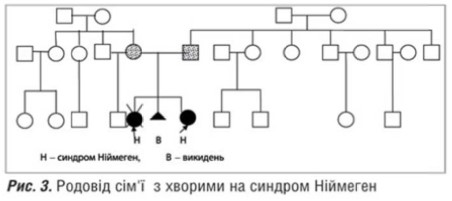
Наводимо більш ретельний аналіз клінікопараклінічниих характеристик двох інших випадків. Зазначимо, що одна із хворих є с ибсом вже згаданої дитин и, що померла у віці трьох років від лімфогрануломатозу, і як повторний сімейний випадок заслуговує на уввагу в першу чергу. Оттже, дитина-пробанд П. (рисс.1,2) народдлась від ттретьої доношеної вагітності у молодиих здорових баттьків, які походять з о дного гірського села, але спорідненістть шлюбу заперечують. На час народження дитинни батькові було 28, маатері — 24 рокии. Від першоої вагітностіі народилась дитина з синддромом Ніймеген; друга вагітністть закінчилась мимовільним викиднемм у ранньому термміні (ммал.3). Вагітність пробандом перебігала із загрозоою перериваання, з привводу чого було проведено лікування. За даними УУЗД виявленно маловоддя. Дівчинка народилась від терміновихх пологів з массою тіла 28000 г, довжиною 47 см, окружністю голівки 30 см, що на три сигмальні відхилення меннше норми відноссно антропометричних показників (россту і маси) ддтини і вииходило за третій центиль нормативів окружністю голівки для новонароддженної дитини. Фенотипові осообливості пацієнта: спплющене чоло, легкий екзофтальм, гіпоплазія нижнньої щелепи, відносно великий ніс і виступаюча середння частина обличчя, епікант, диспластичні вууні раковини. Обтяжений генеалогічниий анамнез та наведені вище особливості фенотиипу дозволили з високою віроогідністю припустити синдром Ніймегеен у даної ддитини. В результаті п роведеного обстеження було виявленно: в гемограмі тенденція до лейкопеннії: кількість лейкоцитів коливалась від 33,1 до 5,2х109 та лімф опенії < 1,00х109. Постійно зниженими ббули lgA — 5–10 мг/1000 мл, різко знижжений рівеньь lgG (до першої заміснної інфузії в нутрішньовенного імуноглобуліну йогоо вміст відповіддав 80 мг/1000 мг, в подальшому коливався відд 23 ддо 320 мг/1000 мл. Спостерігались також змінни з боку клітиннної ланки імунітету: ССД3+ 17–446%, СД4+ 8–22%%, СД8+ 188–19% (в ммжах норми); співвідношення СД4/СД88 — 0,9; ріввень СД19 ппостійно був зни женим — відд 5 до 8%.
Таким чином, іммунологічні показники свідчаать на користь важкого комбіноваано імунодеефіцииту. При проведенні інструментальних обстежень було виявлено гіпоплазію правої ниркии зі збереженою її структурою; з боку іншших внутрішшніх органів патології не вияввлено. Проведення молекулярнно-генетичного обстеження виявило слов’янську мутацію NBBS1 гена — 657deel5 в гомозииготному стаані у пробаннда таа гетерозигоотному станіі у матері. Дитина знаходитться на реггулярній заммісніій терапії внутрішньовенним імуноглобуліномм в дозі 0,2–0,4 гг/кг (веденння препаратту проводиться в стаціонарниих умовах щомісячно). На фоні заммісноої терапії іммуноглбулінном в поєдннанні з антиоксидантною та антибакттеріальною терапією (при появі інфекційних ускладднень) стан дитини клінічно стабільний.
. (рис.4),
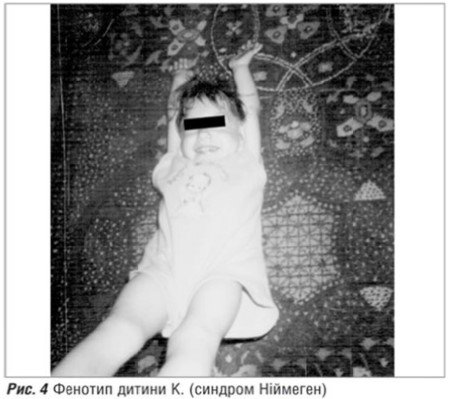
Другий випадок цікавий тим, що у дитини К. (рис.4), м.. Івано-Франківськ, наявні також харакктерні клініко-фенотипові особливості, що приитаманні синдрому Ніймеген. В той же час найбільш поширену мутацію гена NBS1 не виявленоо. Дані генеалогічного аналізу такі: батьки здорові, спорідненість шлюбу заперечують; батько дитиини служив на радіолокацційній станції, працює маляром-штукатуром, оттже, має шкідливі умови праці — контаккт з маслян имитта ацетоновими фарбнииками. Під час вагітності у матері ГРВІ, загроза переривання , хронічна плацентарна недосттатність. Дитина народилася від передчасних полоогів в 34 тижні гестації, з массою тіла 24000 г, довжинною 44 см, окружністю голівкки 28 см, що на 2,8 сигмальні відхиленння менше норми для даного термміну гестації. В періоді новонародженості дитина знахоодилась у відділенні паттології недоношених дітей з наступнимии діагнозами: вроджена мікроцефалія з аномальною структуроою ЦНС, веетрикулодилатаацією; вроджжена пневммонія, ускладнена синддромом легеневої гіпертензії; ДН ІІ ступеня; гіпоксично-ішемічна кардіопатія; гіпогаммаглобулінемія новонародженої дитини. Таким чином, уже в періоді новонародженості у дитини були наявні основні ознаки синдрому Ніймеген
Враховуючи поєднання мікроцефалії із значною затримкою в рості (-4σ), характерними лицьовими дизморфіями, часті ГРВІ і пневмонії, у дитини з високою імовірністю встановлено діагноз синдрому Ніймеген у шестимісячному віці.
На користь важкого імунодефіциту свід чать також перенесена бактеріальна деструкція легень з лівобічним тотальним пневмотораксом у віці 8 місяців. За даними імунологічного обстеження у дитини діагностовано комбіновану форму імунодефіциту. В останні 6 місяців дівчинка щомі сячно отримує замісну терапію внутрішньом‘оязвим імуноглобуліном у зв‘язку з вираженою алергічною реакцією на внутрішньовенний імуноглобулін. В даний час стан дитини відносно компенсований, з ініціативи матері її влаштовано в спеціа лізований заклад для дітей із проблемним інтелектуальним розвитком, хоча є небажаним перебування дитини з важким імунодефіцитом в органі зованому колективі.
Висновки
Синдром Ніймеген є одним з найпоширеніших спадково зумовлених синдромів імуно- дефіциту в нашій популяцій і відрізняється як певним клінічним поліморфізмом, так і генетичною гетерогенністю.
Необхідне подальше удосконалення діагностичних програм, спрямованих на виявлення рідкісних форм мутацій гена даного захворювання та вирішення питання пренатальної його діагностики.
Література
Дранник Г.Н. Клиническая иммунология и аллергология. — Одесса: Астро Принт, 2003.
Иммунодефицитные состояния / Под ред. В.С. Смирнова, И.С. Фрейдлин. —СПб.: Фо- лиант, 2000.
Імунологія: Підр. / А.Ю. Вершигора, Є.У. Пастер, Д.В. Комебота ін.; За ред. Є.У. Пастер. — К.: Вища школа, 2005.
Клиническая имунология и аллерголо- гия: Учебн. пособ. / Под. ред. А.В. Караулова. — М.: МИА, 2002. — 65с.
Наказ МОЗ України від 09.07.04 р. №355
«Про затвердження Протоколів лікування дітей за спеціальністю «Дитяча імунологія».
Чернишова Л.І., Самарін Д.В. Первинні комбіновані імунодефіцити у дітей. — К., 2004 — 240с
3. Діти з синдромом мікроцефалії, як група ризику по синдрому Ніймеген, заслуговують рете— льного імунологічного та молекулярно— генетичного обстеження з метою раннього вияв— лення у них даного захворювання та призначення замісної імунотерапії.
.



Комментировать