Влияние уровня компенсации на прогноз при СД 2. обзор исследований и литературы.
Мохорт Т.В.
Белорусский государственный медицинский университет
Сахарный диабет 2-го типа (СД 2), является единственным из неинфекционных заболеваний, рост заболеваемости которым и негативное влияние на здоровье обусловили принятие резолюции ООН, актуализирующей мероприятия по профилактике, своевременной диагностике и адекватному лечению этой патологии. Независимо от уровня развития медицины и используемых терапевтических подходов, смертность от сердечно-сосудистых осложнений при СД превышает показатели общей популяции [4, 6, 7]. СД 2 является факто- ром риска ампутаций, слепоты и почечной недостаточности, что предопределяет необходимость проведения адекватной гипогликеми- зирующей терапии.
Известно, что повышение уровня гликозилирован- ного гемоглобина (НbА1) на 1% ассоциировано с повышением риска сердечно-сосудистых заболе- ваний на 18% [32], что предопределяет целесооб- разность интенсивной гипогликемизирующей те- рапии с достижением близких к нормальным зна- чений гликемии. В 1970 г. были опубликованы ре- зультаты исследования Диабетической программы университетской группы (UGDP), свидетельствующие что снижение уровня тощаковой гликемии со 170–186 мг/мл до 130–146 мг/мл не обеспечивает достоверного снижения риска сердечно-сосудистых исходов [23]. Практически аналогичные результаты были получены позднее в исследованиях United Kingdom Prospective Diabetes Study, Kumamoto, Veterans Affairs Diabetes Trial (VADT), Action to Control Cardiovascular Risk in Diabetes (ACCORD), Action in Diabetes and Vascular Disease: Preterax and Diamicron Modified Release Controlled Evaluation trial (ADVANCE), в которых более жесткий контроль гликемии не ас- социировался с достоверным снижением риска сердечно-сосудистого прогноза [1, 16, 27, 41]. Ре- зультаты метаанализа перечисленных исследо- ваний по оценке влияния на сердечно-сосудистый прогноз представлены в табл. 1. В качестве при- чин отсутствия снижения риска макрососудистых осложнений анализируются в основном 2 направления: негативные влияния гипогликемических эпизодов и необходимость мультифакториальных влияний с учетом обязательного использования гипотензивной терапии и коррекции липидного профиля крови [17].
Влияние уровня компенсации на прогноз при СД 2 [36]
Исследо- вание | Год | Коли- чество пациен- тов | Интен- сивный контроль гликемии | Стандартный контроль гликемии | Дли— тель- ность | Снижение от- носитель-ного риска сердеч- но—сосудистой смертности | P |
| ACCORD | 2008 | 10 251 | <6% | 7–7,9% | 3,4 | +35% | 0,02 |
| ADVANCE | 2008 | 11 140 | ≤6,5% | >6,5% | 4,9 | –12% | 0,12 |
| UKPDS | 1998 | 3867 | <6 ммоль/л | Максимально достижимый уровень только на диете | 5,0 | +2% | Нет данных |
| VADT | 2008 | 1791 | 6,9% | 8,4% | 5,6 | +32% | 0,26 |
В то же время, доказано, что гипогликемические реакции являются лимитирующим фактором в достижении длительной нормогликемии, так как вызывают постгипогликемические гипергликемии и другие негативные эффекты [15]. При наличии связи СД с поражениями почек, периферической нервной системы, сетчатки вопрос о влиянии СД на когнитивные функции остается сравнительно мало изученным. Спорным вопросом является степень вклада различных факторов (гиперглике- мии, сосудистой патологии, гипогликемии, инсу- линорезистетности) в развитие когнитивной дис- функции при СД. И, наконец, определено негатив- ное влияние гипогликемичеких эпизодов на сер- дечно-сосудистый прогноз.
Целевой уровень НbА1с, согласно действующему консенсусу Европейской ассоциации по изучению диабета и Американской диабетической ассоциа- ции, составляет менее 7,0% (при нормальных значениях менее 6,0%). При этом указывается, что у пациентов с сердечно-сосудистой патологи- ей или наличием в анамнезе гипогликемических эпизодов целевой уровень НbА1с – менее 7,5%, а у пациентов «низкого риска» без сердечно- сосудистой патологии и гипогликемиче-ских эпи- зодов – менее 6,0%. Аргументом для разграничения целевых показателей компенсации СД является повышение риска сердечно-сосудистых событий, ассоциированного с достижением низких зна- чений НbА1с [25]. Критерием гипогликемии являе- тся уровень глюкозы крови мене 3,9 ммоль/л при домашнем мониторинге гликемии с калибровкой по плазме крови [2]. Некоторые исследователи рекомендуют использовать более низкие значения гликемии – 3,5 ммоль/л [3]. Этот параметр по- вергается сомнениям, так как у здоровых людей регистрируются более низкие значения гликемии без клинической симптоматики. Однако именно уровень глюкозы 3,9 ммоль/л инициирует контрре- гуляторный ответ и, следовательно, может сопровождаться клиническими проявлениями.
Изучение влияния гипогликемических эпизодов началось с клинического использования инсули- нотерапии и продолжается до настоящего време- ни. В 1922 г. при начале использования инсулино- терапии было определено, что «пациенты свобо- дны от кетоацидоза, но не свободны от сахара» и было доложено о снижении памяти и внимания при проведении когнитивного тестирования. По данным последних исследований подтверждено, что острая гипогликемия провоцирует психологические нарушения, вызывающие сердечно-сосудистые кризы и гематологические нарушения посредством активации симпатоадреналовой сис- темы и дисрегуляции гормональной системы, направленных на протекцию повреждения мозга.
При этом гемодинамические и гематологические нарушения, развивающиеся на фоне эндотелиальной дисфункции, характерной для СД, повышают риск локальных очагов тканевой ишемии и ма- нифестных сосудистых событий, а сосудистые по- вреждения включают гемореологические наруше- ния, активацию лейкоцитов, вазоконстрикцию, ак- тивацию провоспалительных цитокинов и медиа- торов воспаления [38].
Считается, что при использовании препаратов су- льфонилмочевины 10% зарегистрированных сме- ртельных исходов обусловлены гипогликемическими эпизодами различной степени тяжести [14].
По мнению C. Nordin, гипогликемические эпизоды являются доказанным фактором риска развития острых сердечно-сосудистых катастроф, что свидетельствует о необходимости компенсации гликемии без гипогликемических эпизодов для сни- жения рисков сердечно-сосудистой летальности. Есть мнение о проаритмогенном влиянии гипогли- кемических реакций за счет непосредственного влияния гипогликемии и катехоламиновых реак- ций, вызывающих гипокалиемию и другие нарушения [26].
По данным Veterans Affairs Diabetes Trial (VADT) определено, что выраженные гипогликемические реакции при СД 2 являются одним из основных предикторов инфаркта миокарда, мозгового инсу- льта и смерти от всех причин [16]. Отправным моментом для планирования крупномасштабного многоцентрового рандомизировнного двухфакто- риального исследования ACCORD (Action to Control Cardiovascular Risk in Diabetes) являлось поддержание компенсации сахарного диабета с обеспечением уровня гликозилированного гемоглобина (НbА1с) менее 6,5%, так как в патогенезе ангиопатий, протекающих с поражением как мелких, так и крупных сосудов, доказана первоочере- дная роль гипергликемии.
Но в итоге исследование ACCORD, в котором на- блюдались 10 251 пациентов высокого риска с СД 2, рандомизированных на группы интенсивного (НbА1с менее 6%) и традиционного (НbА1с 7,0–7,9%) гликемического контроля с различной интенсивностью контроля артериального давления и липидного профиля, было прекращено досрочно. Причиной досрочного прекращения исследования ACCORD явилась высокая смертность в группе интенсивного лечения (257 смерти, средний HbA1c 6,4% против 203 смертей, средний HbA1c 7,5% в группе традиционного лечения, т.е. 54 избыточные смерти в группе интенсивной терапии) [29].
Углубленный анализ результатов этого исследования свидетельствует, что количество зарегистрированных эпизодов гипогликемии, требующих медицинской помощи, было значимо выше в груп- пе интенсивного гликемического контроля (10,5% vs 3,5%), независимо от количества зарегистриро- ванных эпизодов [19] (рис. 1). Так, 1 гипогликеми- ческий эпизод в группе интенсивного контроля был зарегистрирован у 7,8% пациентов против 2,5% в группе сравнения, а от 3 до 5 эпизодов – у 0,8 и 0,2% больных соответственно.
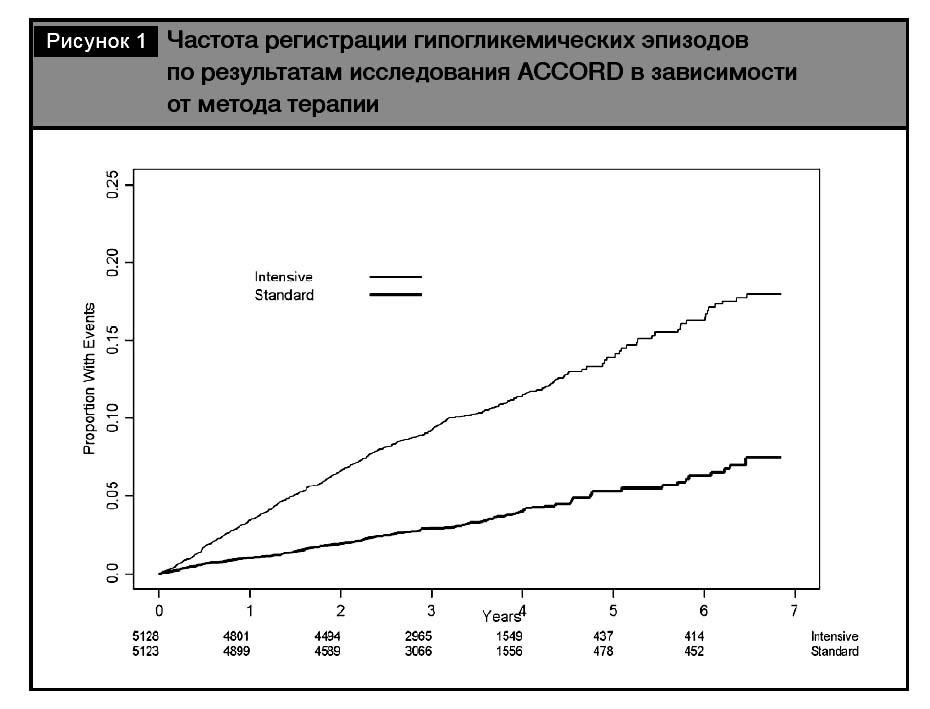
При анализе общего показателя смертности отмечено, что в общей группе пациентов с СД 2 у больных без гипогликемических эпизодов он составил 1,2% в год, а при наличии гипогликемий –3,3% в год. Более того, в обеих ветвях исследования смертность была выше в подгруппах с зарегистрированными гипогликемическими эпизодами (2,8% vs 1,3% в год в группе интенсивного контроля и 4,9% vs 1,1% в год в группе стандартного контроля). Таким обазом, смертность была выше у пациентов с зарегистрированными гипогликемическими эпизодами независимо от терапевтической стратегии. При оценке риска определено, что риск смерти оказался выше в группе интенсивной гипогликемизирующей терапии у лиц без зарегистрированных гипогликемических эпизодов, в то время как в группе стандартной терапии смертность была максимальной у пациентов с зафикси- рованными гипогликемиями [19]. Сделано заключение, что именно гипогликемии определяют повышение риска негативных кардиальных исходов.
В исследовании NICE-SUGER также продемонстрировано, что поддержание более жесткого контроля гликемии (целевой уровень 4,5–6,1 ммоль/л) в условиях отделений неотложной терапии сопровождалось увеличением показателя смертности [35].
В то же время в исследовании ADVANCE (Action in Diabetes and Vascular Disease: Preterax and Diamicron Modified Release Controlled Evaluation trial), по-строенном по схожему дизайну, не было зафиксировано повышения риска инфаркта мио- карда, инсульта, смерти от макроваскулярных событий и смерти от всех причин в группах стандартной и интенсивной тарпии (НbА1с 6,5% при последнем визите). При этом в качестве гипогликеми- зирующего агента в группе интенсивной терапии использовался гликлазид модифицированного высвобождения, что обеспечило крайне редкую регистрацию выраженных гипогликемических эпи- зодов (0,007 против 0,004 событий на 1 пациента в год, p < 0,001) и, тем не менее, не привело к до- стоверному снижению риска сердечно-сосудистых макрососудистых событий и кардиоваскулярной смертности, хотя положительную тенденцию нельзя не отметить. В исследовании ADVANCE ежегодная частота макрососудистых событий (2,2%) была ниже, чем ожидаемый показатель 3,0%, основанный на предыдущих исследованиях СД 2,возможно, вследствие более широкого при- менения статинов, антигипертензивных и антит- ромботических препаратов [2, 42].
Несмотря на то что полученные результаты могут свидетельствовать о том, что понижение глюкозы крови до среднего уровня НbА1с 6,5% в результате лечения не уменьшает риск макрососудистых событий, эти данные не умаляют пользу, которую можно извлечь в результате достоверно получен- ного различия между группами интенсивного и стандартного контроля в снижении риска совокуп- ных исходов – серьезных макро- и микрососудистых событий в группе интенсивной терапии.
Факт связи СД и внезапной смерти подтвержден в различных исследованиях, позволяющих сделать заключение, что СД является независимым фактором риска внезапной смерти и причиной пролонгации интервала QT [10, 28]. С другой стороны, при СД значимо повышается риск кардиомиопа- тий, в патогенезе развития которых имеют значе- ние митохондриальная дисфункция, активация оксидации жирных кислот и другие патологичес- кие процессы, активность которых зависит от ак- тивности клеточного метаболизма и нарушений утилизации глюкозы, сопутствующих гипогликеми- ческим эпизодам. Следствием кардиомиопатиии является развитие нарушений сократительной способности миокарда, усугубляющейся в услови- ях метаболических нарушений и дефицита макроэргов.
Cуществует аргументированная точка зрения, свидетельствующая о том, что при СД регистри- руется удлинение интервала QT на электрокарди- ограмме, определяющее повышение чувствительности миокарда к аритмогенным воздействиям, включая перегрузку кальцием [10, 26] Именно перегрузка кальцием, обусловленная снижением ак- тивности Na-K-АТФазы, в этой ситуации опреде- ляет изменения в саркоплазме и миоплазме, приводящие к нарушениям соотношения внутрикле- точного натрия и кальция и формирования элект- рогенных импульсов в миокарде. С другой сторо- ны, механизм экзоцитоза инсулина связан с бло- кадой калиевых каналов (особенно Ikr и Iks), сопро- вождается нарушением формирования электри- ческих импульсов. Механизм активации секреции инсулина заключается в селективной стимуляции специфических рецепторов на поверхности бета- клетки поджелудочной железы, закрытии АТФ- зависимых калиевых каналов (К+АТФ-каналов), деполяризации мембраны клетки, открытии кальциевых каналов и накоплении ионов кальция в клетке с последующим экзоцитозом инсулина.
Приведенные факты объясняют потенциальный механизм влияния СД 2 на сердечно-сосудистый прогноз, так как блокада АТФ-зависимых калиевых каналов в различных тканях, в том числе клетках миокарда и гладкомышечных клетках и открытие кальциевых каналов при наличии избыточной секреции инсулина и инсулинорезистетности могут быть причиной исходной перегрузки кальцием. Кроме того, имеет значение нарушение барорефлексов, бусловленное присоединением кардиальной автономной нейропатии.
Модель, объясняющая потенциальный механизм внезапной смерти приведена на рис. 2.
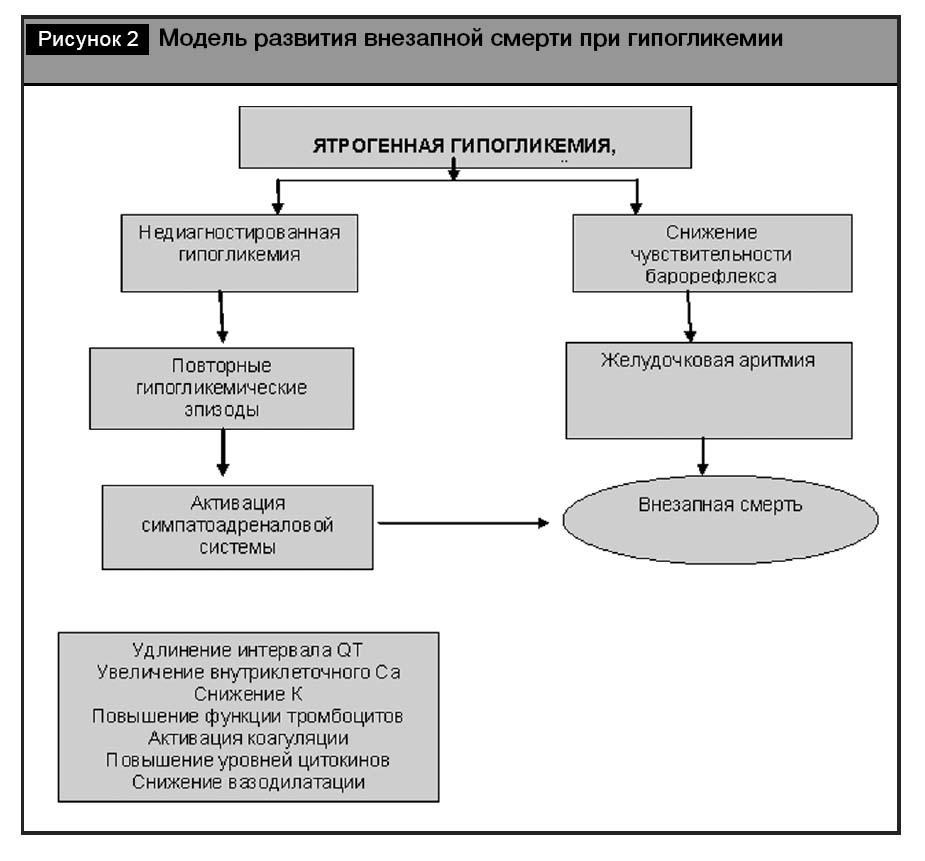
В то же время влияние гипогликемии на повышение риска сердечных аритмий в настоящее время не подтверждено в рандомизированных клинических исследованиях [26]. Изучение роли гипогликемии в повышении риска смерти от сердечно-сосудистой патологии осложняется отсутствием специфических анатомо-морфологических маркеров гипогликемии и тем фактом, что при появлении острых кардиальных болей пациент не всегда определяет уровень гликемии и/или фиксирует постгипогликемическую гипергликемию. Именно перечисленные факторы ограничивают возможно- сти проведения анализа уже имеющихся исследо- ваний и осложняют формирование дизайна исс- ледования, способного доказать или опровергнуть влияние гипогликемий на риск развития аритмий.
Биологические эффекты гипогликемии, определяющие риск сердечно-сосудистых событий, включают контринсулярный ответ на гипогликемию, включающий избыточную секрецию катехо- ламинов и соответствующие кардиальные адренергические эффекты, включая повышение риска аритмий сердца. В настоящее время проаритмогенный эффект гипогликемии, который реализуется в развитие желудочковых аритмий и внезапную смерть, подтвержден как электрофизиологическими, так и единичными клиническими исследованиями на малых группах пациентов.
С учетом вышеприведенных фактов, Американская ассоциация диабета опубликовала данные, что поскольку более половины американцев име- ют уровень НbА1с 7%, а преимущества жесткого контроля сомнительны, целевой уровень НbА1с должен составлять менее 7,0% и может быть определен с учетом оценки риска гипогликемических эпизодов и сердечно-сосудистого прогноза. Для пациентов «низкого риска» без кардиальной патологии целевой уровень НbА1с должен соста- влять менее 6,0%, а у лиц «высокого риска» с се- рдечно-сосудистыми заболеваниями в анамнезе и зарегистрированными гипогликемическими эпизодами – менее 7,5% [2].
Вторым важнейшим фактором, огра-ничивающим снижение уровня НbА1с до нормальных значений, является наличие связи гипогликемий, когнитив- ных нарушений и деменции (или постгипоглике- мической энцефалопатии) [20, 30, 37]. Определе- но, что индекс гипогликемий нарастает с длительностью СД более 6 лет, при этом у больных вы- раженные гипогликемии ассоциированы с высоким риском деменции. В 2009 г. опубликованы ре- зультаты наблюдения за когортой 16 667 пациен- тов с СД 2, наблюдавшейся в течение 27 лет с ре- гистрацией гипогликемических эпизодов [39]. Хотябы о дин гипогликемический эпизод был зарегистрирован у 8,8%. При оценке связи гипогликемиче- ских эпизодов и риска деменции определено, что повторные эпизоды гипогликемии повышают риск деменции. Атрибутивный риск деменции между лицами без гипогликемических эпизодов и с зарегистрированными эпизодами составил 2,39% в год [40]. Многочисленные публикации свидетельству- ют, что при СД 2 отмечаются снижение скорости психомоторных реакций, нарушение функции лобной доли, снижение вербальной памяти, компле- ксные моторные нарушения, визуальные задержки, снижение внимания и др.
Когнитивные и нейрональные нарушения при СД могут быть обусловлены прямым влиянием ятрогенных гипогликемий (глюкоза менее 3 ммоль/л), другими метаболическими нарушениями (метаболический компонент деменции), нарушениями перфузии мозговой ткани (сосудистый компонент деменции), ассоциации с депрессивными состояниями и другими факторами.
Для доказательства клеточных нарушений в экс- перименте на крысах вызывалась острая гипогли- кемия и проводилась оценка функции гиппокампа с использованием мониторинга постсинаптическо- го потенциала и уровня никотин-амид-аденин- динуклеотида (НАДФ) методом флуоресценции. Результаты свидетельствуют о снижении функции гиппокампа и влиянии гипогликемии на память [5,8, 14, 24, 31]. Доказано, что острые и повторные гипогликемии снижаю уровень гликогена в церебеллуме, коре и гипоталамусе на 50% по сравне- нию с нормальными значениями. После остройгипогликемии уровень гликогена возвращается к нормальным значениям через 6 ч, а при повтор- ных гипогликемиях возращение уровня гликогена происходит через 24 ч, что связывают с нарушением контррегуляторных механизмов компенсации гиперинсулинемических гипогликемий [29]. Есть мнение, что гипогликемия инициирует про- граммированную гибель или повреждение нейронов за счет биоэнергетической дисфукнции мозга [3, 34, 39, 40].
В настоящее время определены возможности ви- зуализации мозга для оценки патологических на- рушений, вызванных гипогликемиями. Для этой цели используются методики, основанные на регистрации нарушений метаболизма глюкозы и обеспечения кислородом (позитронно-эмисионная томография, магнитно-резонансная томография и использование различных специальных зондов), позволяющие выявить патологические очаги.
Таким образом, гипогликемические эпизоды ока- зывают негативное влияние на течение СД не только с позиций сложности достижения компенса- ции, но вызывают повышение риска острых сердечно-сосудистых событий и когнитивных нарушений. Сердечно-сосудистые события увеличи- вают вероятность летального исхода, а когнитив- ные нарушения приводят к снижению качества жизни, обуславливают сложности в обеспечении компенсации СД и инициируют развитие микросо- судистых осложнений, также определяющих негативный прогноз [9, 12, 13].
Приведенные факты диктуют необходимость ис- пользования в лечении СД 2 препаратов с низким риском гипогликемических реакций.
Считается, что в 75% случаев смерть при СД 2 является результатом сердечно-сосудистых со- бытий (инфаркта миокарда, аритмии сердца или инсульта), причиной которых может явиться гипогликемический эпизод. Исходя из вышесказанно- го, лечение СД 2 должно планироваться с учетом выбора гипо-гликемизирующих препаратов, гара- нтирующих низкий риск гипогликемических эпизодов [18, 33].
Согласно действующему консенсусу между Европейской ассоциацией по изучению диабета и Аме- риканской диабетологической ассоциацией по лечению СД 2 в качестве препаратов первой линии рекомендуется назначение метформина. Использование в качестве препарата первого выбора инсулинсетитизатора метформина обосновано пато- генетиче-ским влиянием на инсулинорезистетность и низким риском гипогликемических эпизо- дов [11]. Препаратами второй линии являются се- кретагоги – производные сульфонилмочевины и препараты инсулина. Действующие рекомендации определяют целесообразность использования препаратов, обеспечивающих низкий риск гипог- ликемических эпизодов, поэтому в качестве реко- мендуемых производных сульфонилмочевины определены гликлазид и глимепирид, а в качестве препаратов инсулина пролонгированного дейст- вия предпочтение должно быть отдано беспиковым аналогам инсулина гларгину и детемиру.
Следует отметить, что, несмотря на высокий риск гипогликемий при использовании глибенкламида, в течение многих лет ведутся дискуссии о наличии «антиаритмического эффекта» у глибенкламида, объясняемого прямым влиянием препарата на кардиомиоциты, что подтверждается рядом исследований [21, 22].
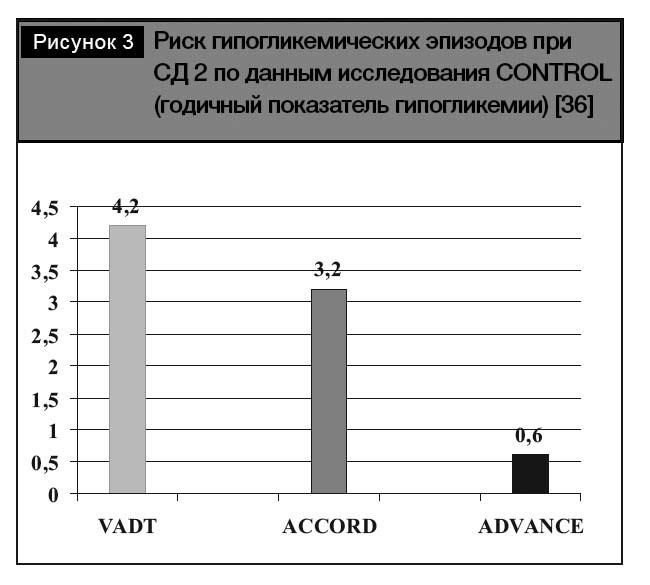
Тем не менее, на рис. 3. приведены результаты сравнения регистрации годичного показателя гипогликемии, свидетельствующего о наиболее низком риске гипогликемических эпизодов при использовании стратегии, базирующейся на назначении преимущественно гликлазида (модифициро- ванного высвобождения), свидетельствующие о минимальной частоте регистрации гипогликеми- ческих эпизодов. Полученные данные позволяют оптимизировать выбор инсулиновых секретагогов с позиций минимизации риска гипогликемий.
В настоящее время достаточно широко используются пролонгированные аналоги инсулина гларгин и детемир, обеспечивающие значимо более низ- кую частоту гипогликемических реакций в течение суток и ночью по сравнению с использованием пролонгированного инсулина (нейтрального про- тамина Хагедорна). В то же время ряд гипоглике- мизирующих препаратов (ингибиторы альфа- глюкозидазы, тиазолидиндионы, инкретины и ингибиторы дипептидил-пептидазы 4, амилин) с низким риском гипогликемических эпизодов отнесены вышеназванным консенсусом, рекомендации которого опубликованы в 2008 г., к препаратам второй линии в связи с отсутствием доказательств негликемических эффектов, реализующихся сни- жением риска развития микро- и макрососудистыхосложнений. В настоящее время наиболее перспективным классом гипогликемизирующих препа- ратов считаются инкретины (экзенатид, лираглю- тид ), так как они позволяют обеспечить достижение целевых уровней компенсации у максималь- ного количества пациентов при отсутствии гипогликемических эпизодов и в связи с наличием обо- снования кардио- и нейропротективных эффектов по результатам экспериментальных и клинических исследований.
ЛИТЕРАТУРА
1. ADVANCE Collaborative Group. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes // NEJM. – 2008. – Vol. 358. – P. 2560–2572.
2. American Diabetes Association. Standards of medical care in diabetes // Diabetes Care. – 2010. – Vol. 33 (Suppl. 1). – S. 1–61.
3. Amiel S.A., Dixon T., Mann R., Jameson K. // Diabet Med. – 2008. – Vol. 25, N 3. – P. 245–254.
4. Antman E.M., Tanasijevic M.J., Thompson B. et al. // NEJM. – 1996. – Vol. 335, N 18. – P. 1342–1349.
5. Auer R. N. // Metab. Brain Dis. – 2004. –Vol. 19. – P. 169–175.
6. Balkau B., Simon D. // Lancet. – 2010. –Vol. 375, N 9713. – P. 438–440.
7. Balkau B. // Lancet. – 1999. – Vol. 353, N 9146. – P. 68–69.
8. Boyle P. J. // Diabetologia. – 1997. – Vol. 40,Suppl. 2. – S69–S74.
9. Bragd J., Adamson U., Bäcklund L.B. et al. // Diabetes Metab. – 2008. – Vol. 34. – P. 612–616.
10. Brown D.W., Giles W.H., Greenlung K.J. et al. // J. Cardiovasc. Risk. – 2001. – Vol. 8. – P. 227–233.
11. Cheng A.Y., Fantus I.G. // CMAJ. – 2005. –Vol. 172. – P. 213–226.
12. Christopher T.K., Elizabeth R. // Endocr. Rev. –2008. – Vol. 29, N 4. – P. 494–511.
13. Cox D.J., Kovatchev B.P., Gonder-Frederick L.A. et al. // Diabetes Care. – 2005. – Vol. 28. –P. 71–77.
14. Cryer P.E. // J. Clin. Invest. – 2007. – Vol. 117, N 4. – P. 868–870.
15. Cryer P.E. // Diabetologia. – 2002. – Vol. 45. –P. 937–948.
16. Duckworth W., Abraira C., Moritz T. et al. // NEJM. – 2009. – Vol. 360, N 2. – P. 129–139.
17. Gaede P., Lund-Andersen H., Parving H-H. et al. // NEJM. – 2008. – Vol. 358, N 6. – P. 580–591.
18. Gerstein H.C., Miller M.E., Byington R.P. et al. // NEJM. – 2008. – Vol. 358. – P. 2545–2559.
19. Ismail-Beigi F., Craven T., Banerji M.A. et al. // Lancet. – 2010. – Vol. 376, N 9739. – P. 419–430.
20. Jacobson A.M., Musen G., Ryan C.M. et al. // NEJM. – 2007. – Vol. 356. – P. 1842–1852.
21. Koltai M. et al. // Acta Physiol Hung. – 1990. –Vol. 75. – P. 175–176.
22. Lomuscio A., Vergani D., Marano L. et al. // Coronary Artery Disease. – 1994. – Vol. 5. –P. 767–771.
23. Meinert C.L., Knatterud G.L., Prout T.E., Klimt C.R. // Diabetes. – 1970. – Vol. 19 (Suppl. 2). –P. 789–830.
24. Miyamoto E. // J. Pharmacol. Sci. – 2006. –Vol. 100. – P. 433–442.
25. Nathan D.M., Buse J.B., Davidson M.B. et al. // Diabetologia. – 2008. – Vol. 51. – P. 8–11.
26. Nordin C. // Diabetologia. – 2010. – Vol. 53. –P. 1552–1561.
27. Ohkubo Y., Kishikawa H., Araki E. et al. // Diabetes Res. Clin. Pract. – 1995. – Vol. 28, N 2. – P. 103–117.
28. Pearson E.C., Woolsey R.L. // Drag Saf. –2005. – 14. – P. 747–753.
29. Pfeffer MA ACCORD(ing) to a Trialist // Circulation. – 2010. – Vol. 122, N 8. – P. 841–843.
30. Ryan C.M., Williams T.M., Finegold D.N., Orchard T.J. // Diabetologia. – 1993. – Vol. 36,N 4. – P. 329–334.
31. Sadgrove M.P., Beaver C.J., Turner D.A. // Brain Res. – 2007. – Vol. 1165. – P. 30–39.
32. Selvin E., Marinopoulos S., Berkenblit G. et al.// Ann. Intern. Med. – 2004. – Vol. 141, N 6. –P. 421–431.
33. Skyler J.S., Bergenstal R., Bonow R.O. et al. // J. Am. Coll. Cardiol. – 2009. – Vol. 53, N 3. – P. 298–304.
34. Suh S.W., Hamby A.M., Swanson R.A. // Glia. –2007. – Vol. 55, N 12. – P. 1280–1286.
35. The NICE-SUGER study investigators Intensive vs conventional glucose control in critically ill patients // NEJM. – 2009. – Vol. 360. – P. 1283–1297.
36. Turnbull F.M., Abraira C., Anderson R.J. et al. // Diabetologia. – 2009. – Vol.52, N11. – P. 2288–2298.
37. Warren R.E., Frier B.M. // Diabetes Obes. Metab. – 2005. – Vol. 7, N 5. – P. 493–503.
38. Wright R.J., Frier B.M. // Diabetes Metab. Res.Rev. – 2008. – Vol. 24, N 5. – P. 353–363.
39. Wright A.D., Cull C.A., Macleod K.M., Hol-man R.R.J. // Diabetes Complications. – 2006. –Vol. 20, N 6. – P. 395–401.
40. Whitmer R.A.,Karter A.J., Yaffe K. et al. // JAMA. – 2009. – Vol. 301, N 15. – P. 1565–1572.
41. UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33) // Lancet. – 1998. – Vol. 352. – P. 837–853.
42. UK Prospective Diabetes Study (UKPDS) Group. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34) // Lancet. – 1998. – Vol. 352. – P. 854–865.
Медицинские новости. – 2011. – №3.


Комментировать