
Сахарный диабет тип 2 у детей. Современные подходы к лечению, к коррекции гипергликемии и инсулинорезистентности у детей с сахарным диабетом II типа. Изучения проблемы сахарного диабета II типа (СД II) в детской популяции. Спектр клинических проявлений при постановке диагноза у детей. Факторы, способствующие нарушению гомеостаза глюкозы при развитии и прогрессировании СД II. выявление клинических и метаболических особенностей проявления СД II у детей и оценка адекватности схем проводимой сахароснижающей терапии. Результаты проведенных исследований.
А.В. Солнцева
Белорусский государственный медицинский университет, Республика Беларусь.
На сегодняшний день актуальность изучения проблемы сахарного диабета II типа (СД II) в детской популяции не вызывает сомнения [5, 9, 25, 29, 39, 41]. Число больных этим заболеванием в разных странах составляет 8–45% общего количества детей с диабетом [7, 9, 10, 29]. За последнее время в нашей республике распространенность этой эндо- кринной патологии среди детского населения увеличилась более чем на 60%.
СД II является гетерогенным заболеванием, разви- вающимся в результате сочетания врожденных и приобретенных факторов [4]. Генетическая составляющая этой эндокринопатии отражена в табл. 1[29]. Первичный генетический дефект, лежащий в ее основе, полностью не раскрыт. Выявленные редкие генетические мутации составляют менее 5% случаев СД II [29]. Очевидно, развитие заболе- вания определяет совокупность множества генов, контролирующих как синтез инсулина островковы- ми клетками, так и его взаимодействие с клетками- мишенями в периферических органах [2].
Таблица 1. Гены-кандидаты, участвующие в регуляции гомеостаза глюкозы у человека и патогенезе СД II [29]
| Факторы, связанные с инсулином | Факторы, секретируемые жировой тканью |
| Рецептор инсулина | Метаболиты |
| Субстраты инсулинового рецептора (IRS-1– |
IRS-4)Жирные кислотыФакторы транскрипцииГлицеролЭнзимы (фосфатазы, серинкиназы, тирозин- киназы, фосфатидилинозитол-3-киназа)Сигнальные протеиныЛептинСпецифические тканевые факторыАдипонектинТранспортеры глюкозы (GLUT4)РезистинГликогенсинтазаАдипсинЭнзимы (фосфоенолпируват карбоксикина- за)Интерлейкин-6, 8К+-каналФактор некроза опухоли-αРецепторы сульфонилмочевиныВсе компоненты ренин-ангиотензиновой системыИнсулиноподобный фактор роста -1, -2Эстрогены
СД II у детей – сложная многофакторная и полигенная патология, возникающая при тесном взаимодействии генетических и средовых компонентов.
Ранней манифестации заболевания в детской популяции способствуют избыточная масса тела и ожирение, низкая физическая активность, употреб- ление высококалорийных продуктов питания, частая распространенность начальных патологиче- ских изменений углеводного обмена, масса тела при рождении менее 2500 г, физиологическая пу- бертатная инсулинорезистентность (ИР), стрессо- вые состояния, урбанизация, определенная этническая принадлежность [5, 8, 11, 12, 15, 20, 23, 25,27–30, 32, 36].
Спектр клинических проявлений при постановке диагноза у детей варьирует от бессимптомной гипергликемии до диабетического кетоацидоза и гипергликемического гиперосмолярного состояния [5,6]. При манифестации СД II у детей сформировано двойное нарушение: сочетание дисфункции β- клеток и сниженной чувствительности перифериче- ских тканей к инсулину (инсулинорезистентность) (табл. 2) [29]. Дисфункция β-клеток проявляется изменением синтеза или превращения инсулина, нарушением его секреции [4]. Периферическая ин- сулинорезистентность выражена относительно и длительное время компенсируется избыточной продукцией инсулина β-клетками (гиперинсулине- мией), поддерживая углеводный обмен в норме. Гиперинсулинемия приравнивается к косвенным признакам ИР и рассматривается в качестве пред- вестника развития СД II у детей [3, 4, 22].
Таблица 2. Факторы, способствующие нарушению гомеостаза глюкозы при развитии и прогрессировании СД II [29]
| Чувствительность к инсулину |
| Нарушение инсулиновой чувствительности |
Инсулинорезистентностьβ-клеточная дисфункцияСекреция инсулина (гипо-, гиперинсулинемия) Дефицит инсулина
Другие факторы
Клиренс инсулина
(Контр)регуляторный ответ (резистин, глюкагон, лептин)
Наиболее клинически значимым при прогрессировании СД II является снижение инсулиновой чув- ствительности мышечной, жировой и печеночной тканей [42]. ИР мышечной ткани характеризуется уменьшением поступления глюкозы из крови в миоциты и ее утилизации в них. ИР жирового депо выражается в уменьшении чувствительности к ан- тилиполитическому действию инсулина, ведущему к накоплению глицерина и свободных жирных кис- лот – источнику образования атерогенных липопротеидов. ИР ткани печени проявляется понижением синтеза гликогена и активацией процессов гликоге- нолиза и глюконеогенеза. В процессе развития СД II, при начинающемся истощении секреторной функции β-клеток и относительном снижении гиперинсулинемии, вначале страдает функция захвата глюкозы мышечной тканью, затем – гликогенсинтетическая способность печени и в последнюю очередь – снижение липолитической функции жировой ткани [4, 22].
Установлена генетическая предрасположенность к развитию ИР [4, 9]. Клинически она реализуется (в виде метаболического синдрома и (или) СД II) только при влиянии на организм ребенка совокуп- ности соответствующих внешних факторов: избы- точного высококалорийного питания, низкой физи- ческой нагрузки, индекса массы тела более 85 пер- центили для данного пола и возраста, начале по- лового созревания, принадлежности к определен- ной расе/этнической группе и др. [25].
Основные метаболические проблемы при СД II разделяют на обратимые и необратимые. Говоря о ИР, к необратимым относятся генетически обусловленные, прогрессирующие рецепторные и пострецепторные дефекты на уровне печени, жировой и мышечной тканей, применительно к дефектам секреции инсулина – генетически обусловлен- ные, прогрессирующие нарушения, способствующие снижению массы β-клеток, связанные с возрастом, наряду с деструкцией β-клеток и дефекта- ми регенерации [4]. К обратимым изменениям при ИР относят глюкозотоксичность и липотоксичность[4].
Роль ранней манифестации у детей СД II и ИР за- ключается в развитии и прогрессировании сердечно-сосудистой патологии и дислипидемии [3, 18, 20,29, 32, 34]. Это определяет актуальность изучения современных подходов к коррекции гипергликемии и феномена ИР при СД II у детей.
Цель нашего исследования – выявление клинических и метаболических особенностей проявления СД II у детей и оценка адекватности схем проводи- мой сахароснижающей терапии.
Материалы и методы
В исследование включено 20 больных (м/д=9/11) СД II в возрасте 12,2 –16,9 года (средний возраст –14,9 ± 1,6 года), находившихся на лечении в Рес- публиканском детском эндокринологическом центре в 2007–2008 гг. У всех пациентов собран анамнез с уточнением срока гестации, роста и мас- сы тела при рождении, наличия нарушений углеводного и липидного обмена у родственников, рассчитан и оценен суточный калораж пациента относительно его фактических потребностей, определен уровень физической активности.
Оценку антропометрических параметров проводили с использованием индексов массы тела (ИМТ), соотношения окружности талии к окружности бедер (ОТ/ОБ). Показатели ИМТ более 97 перцентили для данного возраста и пола рассматривали как ожирение [15, 17, 41, 43]. Величины ОТ/ОБ более 0,85 у девочек и 0,9 у мальчиков указывали на абдоми- нальную форму ожирения. Уровни артериального давления, превышавшие при трехкратном измере- нии 97 перцентиль для пола и возраста, относили к повышенным [1].
Чувствительность тканей к инсулину оценивали по косвенным показателям: базальным уровням инсулинемии и гликемии, расчетным индексам (инсулин базальный/гликемия базальная (И/Г)), малой модели гомеостаза с определением параметра HOMAIR (Homeostatic Model Assessment) [35, 45]. Согласно рекомендациям Американской кардиологической ассоциации (АНА, 2003), у детей базальные значения инсулинемии в норме не превышают 15 мМЕ/л. Интервал значений 15–20 мМЕ/л характерен для пограничной гиперинсулинемии, более 20 мМЕ/л –для высокой [3]. Показатели HOMAIR более 2,77 свидетельствовали о ИР [3, 4]. Степень компенсации углеводного обмена оценивали по уровням гликированного белка – фруктозамина (ФА) (норма до 285 мкмоль/л). Концентрацию общего холесте- рина (ОХ), триглицеридов (ТГ) определяли фер- ментативным, гликемию – ферментативным глюко- зооксидантным методами с помощью наборов реа- гентов «Cormay». В качестве критериев оценки хо- лестеролемии применяли рекомендации Нацио- нальной образовательной программы по холесте- рину для детей и подростков, согласно которым уровни более 5,2 ммоль/л считались высокими [1,3]. Радиоиммунным методом изучали в сыворотке показатели инсулина (наборы ХОП ИБОХ НАНБ) и С-пептида (наборы «Immunotech», Чехия). Опреде- ляли радиоиммунным методом уровни антител к глютаматдегидрогеназе (GAD 65) (норма < 1,0 Ед/мл) (наборы «Immunotech»).
Обработку полученных результатов проводили при помощи пакета статистических программ Statistica 6.0, различия считали достоверными при P < 0,05.
Результаты и обсуждение
Для обследованных больных СД II характерен отя- гощенный семейный анамнез по ожирению (65%). ИМТ более 30 кг/м2 отмечался у обоих родителей в 25%, у одного – в 40% случаев. Более половины детей (55%) имели родственников, страдающих ожирением. Ожирение у родных сибсов наблюдалось у 2 больных. Установлен высокий процент (60%) распространенности СД II среди родственников 1 и 2 степени родства (по материнской линии – у 75% пациентов, по отцовской – у 25%), подтверждающий наследственный характер нарушений углеводного обмена [25].
По мнению ряда авторов, предрасположенность к развитию ИР и СД II у детей развивается внутриутробно [14, 30, 31, 33]. Постнатальное формирование гиперинсулинемии и ИР является отсроченными последствиями задержки внутриутробного развития и макросомии. Макросомия плода рассматривается как результат действия генетических механизмов, направленных на регуляцию секреции фетального инсулина и определяющих чувствительность к нему фетальных тканей [3]. Ее наличие указывает на существовавшую эмбриональную гиперинсулинемию [3].
Согласно гипотезе о «бережливом генотипе», разработанной J.V. Neel [33], развитие ИР представляет компенсаторно-приспособительную реакцию на алиментарный дефицит нутриентов. Ее значение заключается в адаптации и выживании эмбриона при патологическом течении беременности [3, 31]. Модель эмбриональной ИР сохраняется в генети- ческой памяти ребенка и продолжает работать постнатально на накопление энергии при избыточ- ном поступлении калорий. Это приводит к раннему ожирению, нарушению углеводного обмена, дисли- пидемии в детском возрасте [25]. Задержка внутри- утробного развития усиливает апоптоз β-клеток поджелудочной железы, снижая их массу. Умень- шение массы β -клеток является триггером ранней манифестации СД II у детей при наличии генетиче- ской предрасположенности и влиянии внешнесре- довых диабетогенных факторов [4, 22, 25]. Таким образом, для развития СД II прогностически небла- гоприятным является рождение маловесных детей к сроку гестации [3, 30]. Проведенный нами анализ антропометрических показателей при рождении по- казал соответствие средних значений массы тела (3550 ± 150 г) и роста (54 ± 2 см) больных СД II популяционным нормам. Не выявлено случаев рождения маловесных и крупновесных к нормальному сроку гестации детей.
Одним из значимых факторов, участвующих в генезе ИР при СД II у детей, является процесс полового созревания. Согласно результатам нашего иссле- дования, 80% детей с СД II были пубертатного воз- раста и 20% – допубертатного. Для пубертата ха- рактерны физиологическая гиперинсулинемия, снижение чувствительности к инсулину и формиро- вание относительной ИР [8]. Развитие ИР сопро- вождается активацией ростовой оси: повышается концентрация соматотропного гормона и инсулино- подобных факторов роста, снижается уровень свя- зывающего их белка [3].
Избыточная масса тела в подростковом возрасте также способствует усилению физиологической гиперинсулинемии и ИР [3]. В работе M. Wabitsch вы- явлен высокий процент сопутствующих повышенных сывороточных уровней инсулина (67%) и ИР (индекс HOMAIR более 3,8) (79%) у детей с ожирением [44]. Величины базальной гиперинсулинемии и ИР возрастали пропорционально степени ожирения. По мнению некоторых авторов, ожирение в детском возрасте, особенно по абдоминальному типу, является фактором риска ранней манифеста- ции СД II [3, 18, 20, 27–29, 32, 34].
У 10% обследованных пациентов зарегистрировано повышение показателей ИМТ > 90 перцентили, у 45% > 97 перцентили (из них 78% – девочки). Величины индекса ОТ/ОБ у больных СД II на фоне ожи- рения соответствовали абдоминальному типу отложения жира (мальчики – 0,92 ± 0,04, девочки –0,85 ± 0,02), что указывало на повышенный риск формирования других метаболических нарушений (артериальной гипертензии, гиперурикемии, дисли- пидемии и т.д.). Низкий уровень (35%) выявленного нами акантозиса нигриканса как одного из клиниче- ских маркеров ИР и СД II [25] может быть обуслов- лен небольшой выборкой больных. У 6 детей отмечено повышение артериального давления (систо- лическое/диастолическое 135,37±3,1/85,60±1,4 мм рт. ст.). Нами не установлено увеличения уровней ОХ (4,52 ± 0,26 ммоль/л) и ТГ (1,27 ± 0,38 ммоль/л) в группе обследованных.
Уровни тощаковой гликемии при первичном обра- щении колебались от 5,8 до 11,8 ммоль/л (6,6 ± 1,8 ммоль/л). Средние значения базального С-пептида составили 829,8 ± 317,8 пмоль/л и входили в пределы нормы диагностического набора (160–1100 пмоль/л), что свидетельствовало о сохранении функции β-клеток поджелудочной железы у всех больных. Уровни антител к GAD 65 соответствовали 0,61 ± 0,32 Ед/мл. Случаев кетонурии не зарегистрировано.
Исходные показатели базального инсулина варьи- ровали от 13 до 37 мМЕ/л (24,46 ± 9,2 мМЕ/л), у 55% детей отмечалась гиперинсулинемия (норма 3–17 мМЕ/л). Значения индексов И/Г (3,18 ± 1,22) и HOMAIR (6,45 ± 1,20) подтверждали состояние ИР у обследованных. Учитывая патогенетическую связь развития ИР с ожирением, мы оценили показатели инсулина в двух подгруппах больных: с нормальной массой тела и ожирением. У пациентов с ожирени- ем гиперинсулинемия отмечалась в 44% случаев (4/9), с нормальной массой тела – в 62,5% (5/8). При нормальных значениях инсулина независимо от массы тела развитие заболевания связано с нарушением секреторной функции поджелудочной железы. Полученные результаты указывают на со- четанный характер ИР в генезе СД II у детей: связь физиологической (пубертатной) ИР и ИР, обусловленной дисфункцией β-клеток поджелудочной железы.
Протоколы лечения детей с СД II включают индивидуальную и семейную психологическую терапию, изменение образа жизни ребенка с достижением адекватной физической активности и рационально- го сбалансированного питания, обучение пациента и его семьи [16, 27, 29]. Приоритетной целью тера- певтического воздействия является оптимальный гликемический контроль: уровень гликированного гемоглобина < 6,5%, значения тощаковой глюкозы < 126 мг/дл [25]. Однако достижение целевых пока- зателей гликемии у детей представляет сложную задачу, связанную с физиологическими и психоло- гическими возрастными особенностями. По мнению ряда авторов, не более 8–10% детей с СД II достигают оптимальных цифр гликемического контроля без дополнительной медикаментозной поддержки [25, 39, 43].
Сохранение нарушений углеводного обмена в детском возрасте увеличивает риск развития сердечно-сосудистых заболеваний и дисметаболических проявлений диабета [11, 12, 25]. Это определяет долгосрочность дополнительного использования в педиатрической практике фармакологических средств, корригирующих ИР. Для нормализации гликемии у детей при СД II рекомендуется использовать метформина гидрохлорид в виде моно- или комбинированной терапии с инсулином [5, 7,13, 25, 29, 37, 46].
С 2007 г. для лечения детей с 10 лет разрешен к применению препарат «Сиофор 500/850» (мет- формина гидрохлорид, «Берлин-Хеми») [18]. Обычная стартовая доза в детском возрасте со- ставляет 500 или 850 мг однократно. Коррекция до- зы метформина проводится на основании показа- телей гликемии через 10–15 дней до достижения максимальной рекомендуемой суточной дозы пре- парата (2000 мг). Острые обратимые побочные эффекты метформина в отношении желудочно- кишечного тракта можно уменьшить до минимума путем приема лекарственного средства с едой или после еды, а также применением меньших дозировок, которые медленно увеличиваются [5].
Основной механизм действия метформина заключается в снижении продукции глюкозы печенью че- рез активацию рецептора инсулина, как правило, посредством субстрата-2 инсулинового рецептора. В настоящее время влияние метформина на мета- болизм глюкозы оценивается преимущественно как антигипергликемическое действие. Снижение пока- зателей гликемии при использовании препарата не связано с повышением концентрации инсулина в плазме крови. Наблюдаемое снижение массы тела обусловлено анорексигенным эффектом. Отмечается уменьшение жирового депо (больше подкожного, чем висцерального) [25, 29].
Метформин улучшает периферическую чувствительность к инсулину. Снижение периферической ИР приводит к нормализации утилизации и метабо- лизма глюкозы в печени, мышцах и жировой ткани, что предотвращает развитие гипергликемии и кли- нических проявлений СД II у детей.
К настоящему времени завершено три рандомизи- рованных двойных слепых плацебо- контролируемых исследования применения мет- формина у детей с ИР, нормальной толерантностью к глюкозе и наличием отягощенного семейно- го анамнеза по СД II [21, 26, 40]. В первой работе у 13 больных с ожирением и ИР установлено умень- шение по сравнению с плацебо массы тела (- 4,35 кг, Р = 0,02), ИМТ (- 1,26 кг/м2, Р = 0,002), окружно- сти талии (- 2,8 см, Р = 0,003), подкожного жирового депо (-52,5 см2, Р = 0,002) и уровней базального инсулина (-2,2 мЕД/л, Р = 0,011). Чувствительность к инсулину улучшилась у 45% детей на фоне прие- ма препарата и у 27% пациентов группы контроля (Р = 0,21) [40]. Результаты другого исследования (n= 29) показали снижение ИМТ (3,6% относитель- но группы контроля), показателей лептинемии, ба- зальных уровней глюкозы (-0,54 ммоль/л) и инсули- на (-83,3 пмоль/л) у больных даже при отсутствии ограничения пищевого калоража [21]. При изучении влияния метформина у детей с ожирением на фоне низкокалорийной диеты отмечено снижение массы тела на 2,7% относительно контроля, а также кон- центраций лептина, инсулина, глюкозы [26].
Проведенное многоцентровое контролируемое исследование эффективности и безопасности метформина у детей с СД II подтвердило улучшение гликемического контроля через 2 недели от начала терапии со снижением уровней базальной глюкозы на 42,9 мг/дл и гликированного гемоглобина на 1,4% от исходных значений по сравнению с группой сравнения (P < 0,001) [24].
Разработанный нами алгоритм лечения больных с СД II включает соблюдение диеты с суточным ка- лоражем 1800–1900 ккал, ограничением жиров до 25% от общей калорийности и легкоусвояемых углеводов, ежедневную физическую нагрузку, применение сахароснижающих препаратов – бигуанидов (метформин гидрохлорида) и (или) генноинженерного инсулина (пролонгированного и (или) короткого действия) (рисунок).
У детей начальная доза метформина составляла 500 мг 1 раз в сутки после ужина. Назначение препарата проводилось в режиме титрации (по 500 мг) еженедельно. Максимальная доза достигала 2 000 мг в сутки (2–3 приема).
Через 3 месяца монотерапии метформином опре- деляли уровень HbA1c. При сохранении показате- лей HbA1c > 7,0% (или ФА > 315 мкмоль/л) и глике- мии натощак > 6,1 ммоль/л дополнительно назна- чали инсулин пролонгированного действия (инсу- лин НПХ (нейтральный протамин Хагедорна)) или аналоги инсулина пролонгированного действия. Повторно контролировали значения HbA1c через 3 месяца. При отсутствии достижения целевых уров- ней гликемического контроля (базальная гликемия: цельная капиллярная кровь – 3,5–6,1 ммоль/л или плазма – 3,89–7,22 ммоль/л, постпрандиальная гликемия до 8,9 ммоль/л, гликемия перед сном до 6,1 ммоль/л), повышенных показателях гликиро- ванных белков больного переводили на базис- болюсный режим инсулинотерапии (три инъекции инсулина короткого (аналог инсулина ультракороткого действия) действия перед основными приемами пищи и две инъекции инсулина НПХ или аналогов инсулина пролонгированного действия) с отменой метформина.
75% обследованных нами детей находились на монотерапии метформином (500–2000 мг/сут, средняя доза 1356,3 ± 643,8 мг/сут). Перевод на комбинированную терапию (метформин + инсулин (аналог) пролонгированного действия) осуществлен у 20% пациентов ввиду недостаточного контроля глике- мии при лечении метформином. Один больной СД II c синдромом Прадера–Вилли находился на ба- зис-болюсной инсулинотерапии. Установленное нами соотношение схем лечения СД II типа отличается от данных J.H. Silverstein et al., отметивших использование у 44% детей пероральных саха- роснижающих препаратов и у 48% – инсулина [38]. Выявленное различие может быть обусловлено от- носительно малым сроком наблюдения за обследованными больными.
Оценку степени метаболической компенсации проводили ежемесячно на протяжении 12 месяцев наблюдения: показатели ФА колебались от 204 до 435 мкмоль/л на фоне коррекции схем терапии сахароснижающими препаратами. У детей, получающих только метформин, средние уровни ФА составляли 308,9 ± 15,8 мкмоль/л. При комбиниро- ванном лечении показатели гликированного белка соответствовали 253,9 ± 13,6 мкмоль/л, при инсулинотерапии – 269,5 ± 11,0 мкмоль/л и были достоверно ниже значений ФА при монотерапии пероральными препаратами (P < 0,05).
Таким образом, на момент верификации диагноза СД II у обследованных детей выявлен феномен инсулинорезистентности, отражаемый достоверным повышением индексов И/Г и HOMAIR. Степень выраженности описанных нарушений углеводного обмена варьировала индивидуально и была обусловлена сочетанием физиологической (пубертатной) ИР и ИР, обусловленной дисфункцией β-клеток поджелудочной железы. На фоне оптимизации индивидуаль- ного лечебного подхода с применением разных схем терапии отмечено достижение оптимального гликемического контроля по уровням гликированного бел- ка у всех пациентов. Необходимо отметить, что долгосрочным результатом эффективности комплексного лечения СД II у детей должно стать снижение частоты случаев развития и прогрессирования поздних осложнений заболевания, ведущих к ранней инвалидизации и смертности больных.
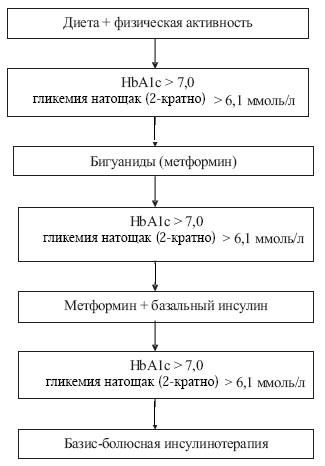
Рис. Алгоритм лечения CД II у детей
Литература
1. Князев Ю.А. Возрастные гормонально-метаболические нормативы: науч.-метод. пособие для педиатров и эндокринологов. – М., 1998.
2. Кураева Т.Л. // Сахарный диабет. – 2005. – № 3. – С. 14 –16.
3. Малявская С.И., Дворяшина И.В., Терновская В.А. Метаболический инсулинорезистентный синдром: диагностика, клиническое значение, педиатрические аспекты. – Архангельск: Сев. гос. мед. университет, 2004.
4. β-клетка: секреция инсулина в норме и патологии / под ред. И.И. Дедова. – М., 2006.
5. Сворен Б.М., Вульфсдорф Д.И. // Intern. Diabetes Monitor. – 2006. – Vol. 18, N 5, 6. – P. 1 – 14.
6. ADA // Pediatrics. – 2000. – Vol. 105. – P. 671–680.
7. ADA consensus statement. Type 2 diabetes in children and adolescents // Diab. Care. – 2002. – Vol. 25. – P.89–94.
8. Arslanian S.A., KalhanS.C. // Diabetes. – 1994. – Vol. 43. – P. 908–914.
9. Aye T., Levitsky L.L. // Curr. Opin. Pediatr. – 2003. – Vol. 15. – P. 411–415.
10. Banerji M.A., ChikenR.I., Huey H. et al. // Diabetes. – 1994. – Vol. 43. – P. 741–745.
11. Caprio S., Tamborlane W.V. // Endocrinol. Metab. Clin. North Amer. – 1999. – Vol. 28. – P. 731–747.
12. Caprio S. // J. Pediatr. Endocrinol. Metab. – 2002. – Vol. 15. – P. 487–492.
13. Castells S. // J. Pediatr. Endocrinol. Metab. – 2002. – Vol. 15 (suppl. 1). – P. 531–540.
14. Cho N.H., Silverman B.L., Rizzo T.A. // J. Pediatr. – 2000. – Vol. 136. – P. 587–592.
15. Cole T.J., Bellizzi M.C., Flegal K.M. et al. // BMJ. – 2000. – Vol. 320. – P. 1240–1243.
16. Daniels S.R. // Curr. Atheroscler. Rep. – 2001. – Vol. 3. – P. 479–485.
17. Deckelbaum R.J., Williams C.L. // Obes. Rev. – 2001. – Vol. 9. – P. 239–243.
18. Eis E.B. // Deutsches Artzteblatt. – 2007. – H.1–2. – S. A64.
19. Eriksson J.G., Forsen T., Tuomilehto J. et al. // BMJ. – 1999. – Vol. 318. – P. 427–431.
20. Eriksson J.G., Forsen T., Tuomilehto J. et al. // BMJ. – 2001. – Vol. 322. – P. 949–953.
21. Freemark M., Bursey D. // Pediatrics. – 2001. – Vol. 107. – E55.
22. Haffner S.M., Miettinen H., Gaskell S.P. et al. // Diabetologia. – 1996. – Vol. 39. – P. 1201–1207.
23. Jaquet D., Gaboriaau A., Czernichow P. et al. // J. Clin. Endocrinol. Metab. – 2000. – Vol. 85. – P. 1401–1406.
24. Jones K.L., Arslanian S., Peterkova V.A. et al. // Diabetes Care. – 2002. – Vol. 25. – P. 89–94.
25. Kaufmann F. R. // Endocrin. Metab. Disorders. – 2003. – Vol. 4. – P. 33–42.
26. Kay J.P., Alemzadeh R., Langley G. et al. // Metabolism. – 2001. – Vol. 50. – P.1457–1461.
27. Kiess W., Galler A., Reich A. // Obes. Rev. – 2001. – Vol. 2. – P. 29–36.
28. Kiess W., Boettner A. // Adolesc. Med. – 2002. – Vol. 13. – P. 181–190.
29. Kiess W., Boettner A., Raile K. et al. // Horm. Res. – 2003. – Vol. 59 (suppl. 1). – P. 77–84.
30. Levy-Marchal C., Jaquet D. // Ped. Diabetes. – 2004. – Vol. 5. – P. 147–153.
31. Lucas A., Fewtrell M.S., Cole T.J. // BMJ. – 1999. – Vol. 319. – P. 245–249.
32. Morrison J.A., Sprecher D.l., Barton B.A. et al. // J. Pediatr. – 1999. – Vol. 135.
33. Neel J.V. // Amer. J. Hum. Genet. – 1962. – Vol. 14. – P. 353–362.
34. Ong K.K., Ahmed M.L., Emmet P.M. et al. // BMJ. – 2001. – Vol. 322. – P. 949–953.
35. Quon M.J. // J. Clin. Endocrinol. Metab. – 2002. – Vol. 87. – P. 650–654.
36. Ranke M. B. Diagnostics of Endocrine Function in Children and Adolescents. – Leipzig: Heidelberg, 1996.
37. Rosenbloom A.L. // Pediatric Drugs. – 2002. – Vol. 4. – P. 209–221.
38. Silverstein J.H., Rosenbloom A.L. // J. Pediatr.Endocrinol. Metab. – 2000. – Vol. 13 (suppl. 1). – P. 1–7.
39. Silverstein J.H., Rosenbloom A.L. // Curr. Diab. Report. – 2001. – Vol. 1. – P. 19–27.
40. Srinivasan S., Ambler G.R., Baur L.A. et al. // J. Clin. Endocrinol. Metab. – 2006. – Vol. 91. – P. 2074–2080.
41. Stolecke H. Nosologische, metabolische und endokrinologische Aspekte der Adipositas. – Berlin; Heidelberg,1997.
42. Stumvoll M., Jacob S. // Exp. Clin. Endocrinol. Diabetes. – 1999. – Vol. 107. – P. 1643–1648.
43. Viberti G., Kahn S.E., Greene D.A. et al. // Diab. Care. – 2002. – Vol. 25. – P. 1737–1743.
44. Wabitsch M. // Eur. J. Pediatr. – 2000. – Vol. 159. – P. 8–13.
45. Wallace T.M., Levy J.C., Matthews D.R. // Diab. Care. – 2004. – Vol. 27. – P. 1487–1495.
46. Zuhri-Yafi M.I., Brosnan P.G., Hardin D.S. // J. Pediatr. Endocrinol. Metab. – 2002. – Vol. 15 (suppl. 1). – P.541–546.
Источник: Медицинские новости. – 2008. – №14.


Комментировать