Патология печени и поджелудочной железы у детей с впервые выявленным сахарным диабетом. Поджелудочная железа. Описание процессов патологии.
М.В.Лобанова, Минский государственный медицинский колледж
Поджелудочная железа (ПЖ) имеет форму трёхсторонней призмы и локализуется глубоко в подложечной области на уровне 10-го грудного –1-2-го поясничных позвонков. Различают головку, тело и хвост железы. Головка ПЖ расположена в петле 12-перстной кишки, она проецируется справа от белой линии на 1,5-3 см выше пупка, а тело и хвост – на 2,5-4,5 см выше пупка слева от белой линии. По передней поверхности головки проходит общий желчный проток, поэтому инфекция из желчных путей легко может проникнуть в панкреас. С возрастом увеличивается вес ПЖ с 2-3 г до 35-47 г и размеры ПЖ: длина с 5,8 до 14,2 см, толщина с 0,38 до 1,5 см, ширина – от 0,9 до 2,0-3,8 см.
ПЖ состоит из железистой ткани и островкового аппарата. В первые 3 года жизни интенсивно увеличивается масса островков Лангерганса, после 4 лет их количество увеличивается медленно, но быстро нарастает масса паренхиматозной ткани ПЖ.
Основная часть ПЖ – экзокринная ткань, только 1-2% всего объёма железы -эндокринная ткань – островки Лангерганса, находящиеся главным образом в хвостовой части железы. 60% островков Лангерганса составляют в-клетки, продуцирующие инсулин, 25% – а-клетки, продуцирующие глюкагон, 10% д-клетки продуцируют соматостатин, остальные 5% клеток островков Лангерганса продуцируют гастрин, тиролиберин и др. пептиды.
У новорождённых детей функция ПЖ развита слабо – образуется мало сока, а активность ферментов и бикарбонатная ёмкость низки. После кормления грудью быстро увеличивается функция ПЖ, достигая максимального уровня к 2-3 годам. Амилолитическая активность ферментов быстро возрастает после введение прикорма и отнятия ребёнка от груди. Липолитическая активность дуоденального содержимого достигает уровня взрослого только к 12 годам. Недостаточная липолитическая активность ПЖ у здоровых детей первых недель жизни компенсируется липазой слюнных желёз и липазой грудного молока [2].
По мнению Белоусова Ю.В. [8, 9], на сегодняшний день определяется тенденция нарастания частоты заболеваний ПЖ у детей, причём распознавание их представляет значительные трудности вне специализированных учреждений, что нередко сопровождается диагностическими ошибками.
Под острым, острым рецидивирующим, хроническим рецидивирующим панкреатитом следует понимать воспалительно-некротический процесс, развивающийся вследствие ферментативного аутолиза ткани ПЖ., что сопровождается «панкреатическими атаками» и проявляется в виде воспалительных изменений ПЖ и вокруг неё, начиная с отёка до деструктивных форм (геморрагического, жирового некроза, гнойного расплавления).
По мнению многих авторов, в большом проценте случаев отёчная форма панкреатита и ограниченный панкреонекроз являются обратимыми заболеваниями.
Хронический панкреатит представляет собой воспалительно-дистрофический процесс ПЖ с последующим её фиброзом и развитием экзокринной и эндокринной недостаточности.
По определению H. Sarles et all [62], хронический панкреатит (ХП) – это хронический воспалительный процесс, сопровождающийся деструкцией экзокринной паренхимы, фиброзом и облигатной деструкцией эндокринного аппарата ПЖ на поздних стадиях заболевания.
H. Spiro [63] определяет хронический панкреатит как повторяющиеся атаки панкреатита, обычно алкогольного генеза, ведущие к прогрессирующему анатомическому и функциональному повреждению ПЖ, которая полностью не восстанавливается. При этом морфологической и функциональной реструкции не наступает, так как после каждой атаки имеет место формирование очагов фиброза. Наблюдается атрофия ацинусов и островков Лангерганса, мощное разрастание соединительной ткани.
Как острый, так и хронический панкреатит является полиэтиологическим заболеванием.
В этиологической сфере, согласно Шабалову Н.П. [39], у детей первое место занимает непосредственное повреждение паренхимы ПЖ инфекционным фактором.
Ведущая роль в генезе острого панкреатита (ОП) принадлежит вирусной инфекции: эпидемический паротит, вирусный гепатит, краснуха, ветряная оспа, корь, грипп и др.
Реже ОП развивается на фоне бактериальной инфекции: туберкулёз, дизентерия, сальмонеллёз и др.
Обращено внимание на возможное развитие ОП у детей на фоне нерациональных вакцинаций. Первоначально происходит сенсибилизация организма, а затем выработка антител к ткани ПЖ. При последующем поступлении аллергена развивается анафилактическая органоспецифическая реакция по типу Артюса с поражением ПЖ.
Белоусов Ю.В. [8, 9] отмечает аутоиммунный характер заболевания с образованием аутоантител к ткани ПЖ. Несмотря на многообразие патологических факторов, у 10-40% больных установить причину не удаётся (идиопатическая форма заболевания).
Морфологические изменения ткани поджелудочной железы носят стойкий характер, сохраняются и прогрессируют даже после прекращения воздействия этиологических факторов и приводят к экзокринной и эндокринной недостаточности. Морфологическим субстратом ХП является отёк, воспаление и очаговые некрозы ПЖ. Нарушение оттока панкреатического сока способствует внутрипротоковой гипертонии, прогрессированию некроза ацинарной ткани с последующими атрофией ацинусов, и интралобулярным и перилобулярным фиброзом органа. В начальной стадии патологический процесс может носить ограниченный характер и локализоваться в какомлибо одном отделе ПЖ. По мере развития заболевания диффузно поражается вся железа [8, 9].
Патогенез, как острого, так и хронического панкреатита, бывает разным (билиарный, алкогольный хронический). Однако на конечном этапе независимо от причин, вызывающих острый и хронический панкреатиты, процесс сводится к аутолизу ткани ПЖ и проникновению (уклонению) панкреатических ферментов в кровь.
Латентная форма хронического панкреатита часто связана с аутоиммунными процессами. Наличие антипанкреатических антител в крови больных, состояние клеточного и гуморального иммунитета указывают на значение иммунологических нарушений в патогенезе хронического панкреатита [8, 9, 32, 35, 39].
Согласно этиологической классификации нарушений гликемии (ВОЗ 1999 г.) на фоне болезни экзокринной части поджелудочной железы, то есть на фоне панкреатита, рассматривается специфический (не основной) тип сахарного диабета [11].
По вышеупомянутой классификации рассматривается.
1. Сахарный диабет (СД) 1 типа (аутоиммунный – 90% и идиопатический – 10%), характеризующийся деструкцией в-клеток и абсолютной инсулиновой недостаточностью.
2. Сахарный диабет (СД) 2 типа – от преимущественной резистентности к инсулину и гиперинсулинемией с относительной инсулиновой недостаточностью до преимущественного секреторного дефекта с относительной инсулиновой резистентностью или без неё.
3. Другие специфические типы диабета:
генетические дефекты в-клеточной функции; генетические дефекты в действии инсулина; эндокринопатии; болезни экзокринной части поджелудочной железы; д-диабет, индуцированный лекарствами, химикалиями (аллоксан и др.); д- диабет на фоне инфекционного фактора; необычные формы инсулиноопосредованного диабета; другие генетические синдромы, иногда сочетающиеся с диабетом.
4. Гестационный сахарный диабет – диабет только во время беременности.
Сложность вышеизложенной классификации иллюстрируется рядом факторов:
1) «В настоящее время выделена подгруппа СД 1 типа, который развивается постепенно, подобно СД 2 типа. В течение 1-2 лет не требуется лечение инсулином, а затем развиваются все классические проявления СД 1 типа с обязательной инсулинотерапией. Название этого подтипа диабета – СД тип 2, который может быть СД тип 1 в ремиссии» [11].
2) Замысловатость инсулинорезистентности при СД 2 типа.
3) Специфические типы диабета вследствие болезни экзокринной части pancreas, диабета, индуцированного лекарствами, химикалиями, инфекционным фактором, можно было бы назвать СД на фоне панкреатита.
4) Группа генетического дефекта вклеточной функции состоит из 5 подгрупп «СД взрослых у молодых» («matury onset diabetes of the young» MODY 1-5 ), «в основе которых лежит генетический дефект и последующая мутация ядерного транскрипционного фактора – гепатоклеточного нуклеарного фактора-4-альфа (1альфа, 1-бетта)» [11], одним словом – патология печени (стеатоз).
5) В группе генетических синдромов, сочетающихся с СД, рассматривается липоатрофический диабет, характеризующийся инсулинорезистентностью и генерализованным отсутствием подкожного жира. Доза инсулинотерапии достигает 2000 ЕД. «Патогенез не ясен. Причина смертельного исхода – печёночная недостаточность»[11].
6) Гестационный сахарный диабет рассматривается только во время беременности.
7) Абсолютно не отражен высокий процент выявления СД в климактерический период (гораздо больший, чем при беременности).
Если раньше считали, что выявление СД 2 типа характерно для лиц старше 40 лет, то на сегодняшний день отмечено, что абсолютная и относительная инсулиновая недостаточность полностью не корректирует с ранним и поздним возникновением СД [21]. В настоящее время наблюдается более частое выявление СД 1 типа в зрелом возрасте и рост заболеваемости СД 2 типа у детей.
J.Kobberling et al, J.Barbosa, С.В.Кудрякова и др. [19, 54] исследовали HLAфенотип у больных СД 1 типа с поздним началом. Анализ полученных данных показал, что распределение HLAантител в этой группе больных аналогичен таковому у больных с ранним началом заболевания. Авторы пришли к выводу, что СД 1 типа, диагностированный как в раннем, так и в позднем возрасте, имеет одни и те же генетические характеристики.
Признана иммунно-вирусная теория развития СД 1 типа. По мнению многих эндокринологов, от начала инфицирования до появления первых клинических симптомов СД может пройти от нескольких недель до полугода, иногда и больше, что связано с разной степенью сопротивляемости организма инфекции и активностью репаративных процессов. При повреждении более 90% β-клеток появляются клинические признаки СД [18].
В сыворотке крови у большинства лиц в доклинической стадии сахарного диабета, иногда за 10-15 лет до манифестации заболевания и на ранних этапах клинического периода, обнаруживаются аутоантитела к антигенам в-клеток, инсулину и различным изоформам глутаматдекарбоксилазы.
Морфологическое изучение препаратов ПЖ больных, погибших вскоре после манифестации сахарного диабета показало наличие воспалительных инфильтратов из активированных лимфоцитов – так называемый инсулит: скопление CD8 (Т-супрессоров и цитотоксических Тлимфоцитов) и CD4 (Т-хелперов), а также Влимфоцитов, макрофагов, NK-лимфоцитов, не выходящих за пределы островков Лангерганса [4-6].
В целом, сахарный диабет является сложным заболеванием, характеризующимся нарушением всех видов обмена веществ, развитием сначала относительной инсулиновой недостаточности, впоследствии – абсолютной, с патологическими изменениями во всех органах и тканях.
Поддержание нормального уровня гликемии обеспечивается деятельностью печени, поджелудочной железы, гипофизарно-надпочечниковой системой и др. Проследим взаимосвязь и взаимозависимость поджелудочной железы и печени. Печень занимает ключевую позицию в поддержании гомеостаза, играет важную роль в адаптационных условиях, поддерживает межорганные и межсистемные связи. В клетках печени происходит гликогеногенез, его депонирование, гликогенолиз с образованием глюкозы (основного источника питания всех органов и тканей), глюконеогенез.
Глюкоза наряду с жирами и белками является источником энергии. В организме человека, весом 70 кг, запасы энергии в виде гликогена (углеводы) составляют 480 г, (400 г – гликоген мышц, 80 г – гликоген печени), что эквивалентно 1920 ккал (320 ккал-гликоген печени, 1600гликоген мышц). Глюкоза, всосавшаяся в желудочно-кишечном тракте, через портальную вену, поступает в печень. Гликоген имеется почти во всех тканях, но в основном он выявляется в качестве депо энергии в печени и мышцах. Гликоген мышц представляет собой скопления, состоящие из отдельных частичек с молекулярной массой 2.107. В печени содержатся как отдельные, так и агрегированные частицы гликогена, их общая масса более 109, в отдельных частицах на 1 г полисахарида приходится 1,1 г воды. Кроме того, в них определяются ферменты, необходимые для синтеза и распада гликогена. Количество гликогена в печени находится в прямой зависимости от приёма и усвоения пищи и составляет у человека около 400 ммоль (65 г) глюкозы на 1 кг ткани. В скелетных мышцах содержится 85 ммоль глюкозы (14 г) на 1 кг мышечной ткани. Количество гликогена в мышечной ткани зависит от физической нагрузки, оно не меняется даже при голодании, но при обильной физической нагрузке в течение 1-2 часов может снизиться до 1 ммоль на 1 кг ткани. У мужчины, массой тела 70 кг, на долю мышц приходится 28 кг гликогена, на долю печени – 1,6 кг [4-6].
Однако нельзя согласиться, что основное депо гликогена содержится в мышцах. Габариты депо печени трудно сравнить с оными в мышцах. Глюкоза, всосавшаяся в желудочно-кишечном тракте, поступает через портальную вену в печень. Именно печень, а не мышцы, играют основную роль в метаболическом процессе, именно печеньявляется основным депо энергии за счёт несоизмеримого кругооборота обмена веществ.
Головной мозг, относящийся к инсулинонезависимым тканям, массой 1400 г потребляет 80 мг/мин глюкозы. Печень способна генерировать глюкозу со скоростью 130% мг/мин. Таким образом, более 60% глюкозы печени идёт на обеспечение нормальной активности центральной нервной системы, причём это количество неизменно при гипергликемии. При гипогликемии (поражение печени и поджелудочной железы) могут наблюдаться полуобморочные состояния, что способствует прогрессированию диабетической энцефалопатии.
Поддержание постоянного уровня гликемии относится к жизненно важным функциям печени и поджелудочной железы. Резервные возможности печени очень велики. Однако, при метаболическом синдроме, состоящего из «смертельного квинтета» (артериальная гипертензия, сахарный диабет, инсулинорезистентность, ожирение, жировой гепатоз стеатоз) на фоне нарушения клеточного метаболизма, обусловленного жировой инфильтрацией гепатоцитов и следственной инсулинорезистентностью, может наблюдаться синдром неустойчивого сахара в крови [22-27, 30].
В нормальных условиях инсулин прежде, чем поступить в периферическую кровь, проходит через портальную вену печени. При чём в ней задерживается 40-50% гормона натощак, при стимуляции глюкозой количество задерживаемого инсулина в печени может достигнуть 8090%. Чем выше секреция инсулина – тем больше его задерживается в печени. Биологический период полураспада инсулина 7-15 минут. Основную роль в его инактивации (до 80%) играет печень [4-6, 21,36]. Глюкагон, секретируемый а-клетками ПЖ, сначала попадает в межклеточное пространство и интерстициальную жидкость, а затем с током крови через портальную вену – в печень. Основное гликогенолитическое действие глюкагона происходит в печени, где он связывается с рецепторами гепатоцитов и активизирует аденилатциклазу, которая переводит АТФ в цАМФ. В периферических тканях глюкагон оказывает липолитическое действие, повышая липолиз, снижая липогенез и белковый синтез. Концентрация глюкагона в портальной вене колеблется от 300 до 4500 пг/мл, тогда как в периферической крови – до 90 пг/мл. Разрушение глюкагона главным образом происходит в печени, менее в почках.
При нормальном состоянии аи в-клеток ПЖ, при нормальном состоянии печени гипогликемия не развивается даже при длительном голодании. При ограниченном поступлении углеводов (глюкозы) уже через 40-48 ч содержание глюкагона в крови возрастает на 50-100% по сравнению с его концентрацией натощак. Это сопровождается снижением инсулинемии, соотношение уровней инсулина и глюкагона уменьшается до 0,4 (в норме –3,0). Повышенное образование глюкагона ведёт к усилению гликогенолиза, глюконеогенеза и уменьшению запасов гликогена. Снижение секреции инсулина стимулирует липолиз, повышенное содержание глюкагона ускоряет образование кетоновых тел [4-6, 21, 36]. Кетоз, повышенное образование кетоновых тел в результате нарушения метаболизма иллюстрирует нарушение жирового обмена с последующим избыточным накоплением жира в печени.
В нормальной печени содержание жира не превышает 1,5% от её массы, и он не обнаруживается при обычном гистологическом исследовании. Мелкие капли жира в гепатоцитах начинают определяться при световой микроскопии, если его количество возрастает до 2-3%, что расценивается как жировая инфильтрация печени (стеатоз). Основные компоненты гепатоцеллюлярных липидов представлены триглицеридами, субстратами для синтеза которых являются жирные кислоты и глицерофосфат. Жирные кислоты поступают в гепатоцит из нескольких источников: из пищевого жира и в результате липолиза жировой ткани. Печёночная клетка способна самостоятельно синтезировать жирные кислоты из ацетилкоэнзима А, особенно при избытке последнего. Источниками глицерофосфата в гепатоците являются глицерин, образующийся при гидролизе липидов, и глюкоза, которая в ходе гликолиза превращается в фосфатидную кислоту, запускающую реакции синтеза триглицеридов. Таким образом, продукция триглицеридов в гепатоците находится в прямой зависимости от содержания в нём глюкозы, жирных кислот и ацетилкоэнзима А [12, 44, 46, 61].
Жир в клетках печени откладывается в результате избыточного поступления в печень свободных жирных кислот (СЖК), снижения скорости окисления СЖК в метахондриях гепатоцитов; избыточного образования и всасывания СЖК в кишечнике; пониженного синтеза липопротеинов разной плотности в самой печени; функциональной печёночной недостаточности, обусловленной заболеванием печени [10, 12, 13, 44].
Накопление жира в гепатоцитах происходит в том случае, если образование триглицеридов превалирует над синтезом липопротеинов и секрецией последних из гепатоцита в виде ЛОНП [44, 56, 57].
В развитие жирового гепатоза могут включаться эндогенные и экзогенные механизмы. К экзогенным факторам относится повышение всасывания продуктов гидролиза липидов и моносахаридов (глюкоза, фруктоза, галактоза), являющихся предшественниками триглицеридов.
К эндогенным патогенетическим факторам относятся:
усиление периферического липолиза (действие алкоголя, никотина, кофеина и др.);
снижение утилизации жирных кислот гепатоцитами;
повышение синтеза липидов;
блокада ферментов, участвующих в синтезе ЛПОНП и их секреции;
дефицит белков в гепатоцитах;
усиление гликогенолиза (повышенное содержание глюкагона);
снижение гликогеногенеза и следственное снижение содержания гликогена в печени на фоне низкого соотношения уровней инсулина к глюкагону.
По мере снижения гликогена в печени и «накопления жира печёночная клетка становится всё более уязвимой и чувствительной к токсическим влияниям» [44, 46, 60, 61].
Выше изложенные данные иллюстрируют взаимозависимость углеводного, липидного, белкового и минерального обмена веществ, взаимообусловленность экзо и эндокринной части ПЖ, взаимосвязь работы печени и поджелудочной железы. Нарушение предоставленного баланса печени и поджелудочной железы поясняет многогранный механизм развития сахарного диабета на базе патологических изменений этих органов.
На сегодняшний день признана иммунновирусная теория развития СД 1 типа. От начала инфицирования до появления первых клинических симптомов СД может пройти от нескольких недель до полугода, иногда и больше, что связа- но с разной степенью сопротивляемости организма инфекции и активностью репаративных процессов [18].
M.A.Sperling отмечает самый высокий уровень заболеваемости СД у детей в двух воз- растных группах: 5-7 лет и в пубертатный период, что объясняет увеличением контакта с инфекци- ей, воздействием гонадотропных стероидов как антагонистов инсулина и эмоциональными стрес- сами, присущими детям этого возраста. Автор об- ращает внимание на сезонность и длинноволно- вую цикличность в заболеваемости СД 1 типа – число больных увеличивается в осенне-зимний период, особенно у детей в возрасте до 6 лет.
По данным В.Г.Баранова, А.С.Страйковой [7], СД 1 типа у детей наиболее часто выявляется в возрасте 3-4, 6-8, 11-14 лет, что авторы связывают с ростом перестройки и уязвимостью гормо- нальной и иммунной системы организма.
На основании долгих наблюдений нами [28] замечена следующая корреляционная связь выявления СД 1 типа с возрастом детей. 1 пик выявления СД зафиксирован у детей в возрасте 1-3 года, 2 пик – в возрасте 5-6 лет, 3 пик – в возрасте 11-12 лет, 4 пик – в возрасте 15-16 лет. Незадолго до выявления СД ребёнок получил вакцинацию или перенёс простудное, инфекционное заболевание (ветряная оспа, эпидпаротит) с обязательной вакцинацией по «выздоровлении». Именно в выше перечисленные возрастные периоды и проводится вакцинация (см. календарь вакцинации).
И.И. Мечниковым в 1900 г. [31] в результа- те открытия цитотоксических антител было показано, что иммунные реакции не всегда носят защитный характер, в целом ряде случаев они могут привести к возникновению весьма серьёзных патологических изменений.
За последние 10 лет календарный план профилактических прививок насытился до предела. Проследим только первый год жизни ребёнка:
- каждому новорождённому в первые сут- ки на 12-ый час после рождения проводится вак- цинация гепатита В (ВГВ-1);
- на 3-4 сутки – БЦЖ, при чём в той же дозировке, как взрослому;
- на первый месяц жизни – первая ревакцинация гепатита В (ВГВ-2);
- в возрасте 3 месяца – коклюшно- дифтерийно-столбнячная вакцина (АКДС-1) и оральная полиомиелитная вакцина (ОПВ-1);
- в возрасте 4 месяца — АКДС-2 и ОПВ-2;
- в возрасте 5 месяцев – АКДС-3, ОПВ-3,
- в 12 месяцев – тривакцина (живая коревая вакцина ЖКВ, живая паротитная вакцина ЖПВ, вакцина против краснухи).
По исследованиям Classen D.C. (48), вакцинация в первые 8 недель жизни связана с повышением риска заболеваемости инсулинозависимым сахарным диабетом на 90%.
Следует отметить, что вакцинация не даёт 100% гарантии от инфекционных заболеваний в естественном варианте, а приносит серьёзный непоколебимый ущерб всей нейро-иммунногландулярно-органной системе новорождённого.
Согласно литературным данным, долгой врачебной практике, нами замечено, что в патогенезе сахарного диабета (как при относительной, так и абсолютной инсулиновой недостаточности) имеет место высокий процент тотального поражения поджелудочной железы с параллельно идущей патологией печени, желчевыводящих путей, жировым гепатозом (стеатозом).
Нами была поставлена цель – разрешить вопрос высокого роста заболеваемости сахарного диабета, предоставив многогранный патогенез этого полиэтиологического заболевания.
Была установлена задача – показать, что одной из важных причин сахарного диабета является тотальное поражение поджелудочной железы (аутолиз ткани) с параллельно идущей патологией печени (стеатоз) и желчевыводящих путей.
Объектом исследования был взят впервые выявленный сахарный диабет у пациентов детского возраста.
Методы исследования
1. субъективный метод (жалобы пациента, анамнез заболевания, жизни). По детской амбулаторной карте (с рождения) прослежено состояние пациентов согласно графику вакцинации; по истории болезни с впервые выявленным СД изучено состояние пациентов на стационарном лечении; по амбулаторной карте диспансерного лечения СД прослежено состояние больных впоследствии;
2. объективный метод (Status praesens);
3. лабораторный метод (общий анализ крови, мочи, биохимический анализ крови);
4. инструментальный метод – ультразвуковое исследование органов брюшной полости (печени, желчного пузыря, поджелудочной железы, почек);
На базе эндокринологического отделения 2-ой детской городской больницы г. Минска в течение четырёх лет набирался материал исследования: 83 пациента с впервые выявленным сахарным диабетом (СД) в возрасте от 1 года до 15 лет (43 девочки, 40 мальчиков).
83 пациента по возрасту разделены на 5 подгрупп (Табл. № 1).
Таблица 1
| Подгруппа | возраст | общее количество | девочки | мальчики |
| 1 | 1-3 года | 11 | 5 | 6 |
| 2 | 4-6 лет | 24 | 11 | 13 |
| 3 | 7-9 лет | 14 | 7 | 7 |
| 4 | 10-12 лет | 14 | 8 | 6 |
| 5 | 13-15 лет | 20 | 8 | 12 |
Проведено субъективное исследование: опрос детей, их родителей, анализ детских амбулаторных карт (со времени рождения) и стационарных историй болезней.
Обращает на себя внимание стереотипность выявления сахарного диабета в детском возрасте. На первый план выступает корреляционная связь возраста детей и текучей «профилактической» вакцинации. Незадолго до выявления СД ребёнок получает комбинированную вакцинацию или переносит простудное, воспалительное заболевание, а через 0-20 дней после «выздоровления» обязательно получает вакцинацию. Впоследствии у ребёнка наблюдается ухудшение сна, раздражительность, частые головные боли, снижение памяти, успеваемости в школе, отсутствие аппетита, диспептические расстройства, похудание. С выше изложенными жалобами прослеживается посещение терапевта, невропатолога, отоларинголога, дерматолога и др. врачей. Пациент получает общеукрепляющие, седативные препараты, витаминотерапию, физиотерапевтическое лечение. Улучшение не наблюдается. При позднем лабораторном обследовании определяется гипергликемия, глюкозурия и ацетонурия. Как правило, в прекоматозном состоянии (кетоацидотическая кома сахарного диабета) ребёнок попадает на стационарное лечение, где назначается и пожизненно проводится инсулинотерапия.
У четырёх детей 5-ой подгруппы СД развился на фоне алкогольной интоксикации (девочка пила шампанское на день рождения, трое мальчиков – пиво).
При субъективном обследовании детей (их родителей) выявлено употребление в большом количестве непригодных для детей продуктов питания (чипсы, кириешки) и напитков (кокакола, пепси-кола, пиво и др.). Следует отметить нарушение режима питания, перешедшее в привычку.
При опросе отмечается сухость во рту, жажда, снижение аппетита, тошнота, горечь во рту, отрыжка, метеоризм, урчание живота, нарушение функции кишечника, похудание, с пятилетнего возраста больные предъявляют жалобы на боли в животе.
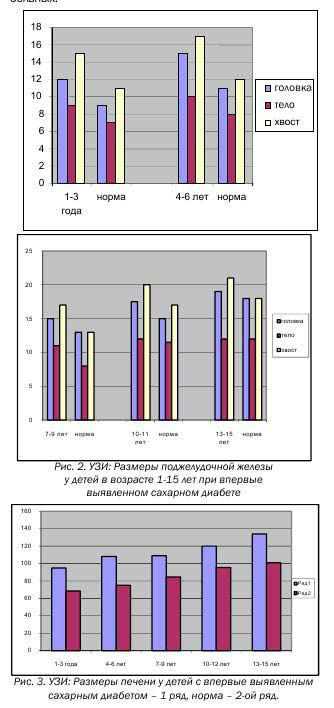
Таблица 2. Размеры поджелудочной железы у детей с впервые выявленным сахарным диабетом 1 типа (данные ультразвукового исследования)
| Возраст | Колво | Размеры поджелудочной железыв мм | ||
| Головка | Тело | Хвост | ||
| 1-3 года | 11 | 11,8 ± 0,9ммнорма 9-10 мм | 8,6 ± 0,6ммнорма 7мм | 14,5 ± 1,0ммнорма 10-11 мм |
| 4-6 лет | 24 | 14,6 ± 1,0ммнорма 11мм | 9,13 ± 0,7ммнорма 7 -8мм | 16,7 ± 1,1ммнорма 12мм |
| 7-9 лет | 14 | 14,9 ± 1,2ммнорма 12-14 мм | 10,6 ± 0,9ммнорма 8 мм | 16,8 ± 1,4ммнорма 13-14 мм |
| 10-12 лет | 14 | 16,7 ± 1,1ммнорма 14-16 мм | 11,2 ± 1,1ммнорма 10-11 мм | 20,9 ± 1,8ммнорма 15-16 мм |
| 13-15 лет | 20 | 18,6 ± 1,7ммнорма 18 мм | 11,0 ± 1,2 ммнорма 12-14 мм | 20,6 ± 1,0ммнорма 18мм |
Возрастные нормативы размеров внутренних органов брюшной полости (поджелудочной железы) у детей в таблице представлены согласно Васильеву А.Ю., Ольховой Е.Б.
При объективном осмотре наблюдается похудание, бледность и сухость кожных покровов, увеличение подчелюстных, шейных, надключичных, подмышечных, паховых лимфатических узлов, безболезненность при пальпации. Обращает на себя внимание увеличение миндалин, их «рыхлость», белый налёт на языке. Следует отметить диффузное увеличение щитовидной железы (у некоторых наличие уплотнений), безболезненность, подвижность при акте глотания. При аускультации – тоны сердца ясные (у некоторых прослушивается систолический шум), тахикардия. При пальпации у всех больных наблюдается гепатомегалия, печень на 1-4 см выступает из-под края правой рёберной дуги, мягкая, эластичной консистенции, чувствительная при пальпации. Болезненность пузырной точки определяется у 4-х больных 2-ой подгруппы, у 4-х больных 3-ей подгруппы, у пяти 4-ой подгруппы, у восьми – 5-ой подгруппы.
Таблица 3. Размеры печени и наличие неправильной формы желчного пузыря у детей с впервые выявленным сахарным диабетом 1 типа
| Возраст | Количество | Желчный пузырь неправильной формы | Размеры печени (правая доля, переднезадний) | Нормальные размеры печени (правая доля, переднезадний) |
| 1-3 года | 11 | 9,1 % | 94,9 ± 1,6мм | 64 – 73 мм |
| 4-6 лет | 24 | 16,7 % | 107,9 ± 1,6мм | 66 – 84 мм |
| 7-9 лет | 14 | 21,4 % | 109,0 ± 1,4мм | 76 – 93 мм |
| 10-12лет | 14 | 28,6 % | 120,6 ± 1,8мм | 95 -96 мм |
| 13-15лет | 20 | 30,0 % | 134,1 ± 0,9мм | 91 – 111мм |
Возрастные нормативы размеров внутренних органов брюшной полости (печени) у детей в таблице представлены согласно данным Е.А. Улезко.
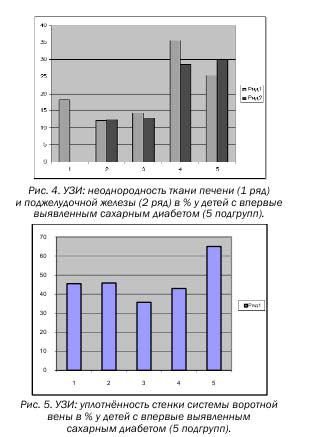
Таблица 4. Изменения со стороны эхогенности ткани печени и поджелудочной железы, наличие уплотнённости стенки системы портальной вены и её размеров
| Возраст | Кол-во | Уплотнённость стенки сосудовсистемыворотной вены | Размеры воротротнойвены | Неоднородность тканипечени | Неоднородность тканиподжелудочной железы |
| 1-3 год | 11 | 45,5 % | 5,4 мм | 18,2 % | — |
| 4-6 лет | 24 | 45,8 % | 6,9 | 12,3 % | 12,5 % |
| 7-9 лет | 14 | 35,7 % | 7,2 | 14,3 % | 12,8 % |
| 10-12 лет | 14 | 42,9 % | 8,1 | 35,7 % | 28,6 % |
| 13-15лет | 20 | 65,0 % | 9,4 | 25,4 % | 30,0 % |
При лабораторном обследовании у всех пациентов – гипергликемия, глюкозурия, ацетонурия, нейтрофильный лейкоцитоз. У детей, переведенных на стационарное лечение в эндокринологическое отделение из инфекционной больницы, прослеживается повышение уровня амилазы.
Ультразвуковое сканирование проводилось на УЗ аппарате «Acuvix» (Корея), работающем в «реальном масштабе времени», использовали конвексный трансдюссер частотой 3,5-5,0МГц.
Обработка выбранного изображения предусматривала возможность получения следующей информации:
1. Увеличение выбранного изображения использовалась для лучшей детализации исследуемого органа.
2. Определение размеров и площади интересующего объекта осуществлялась с помощью электронной линейки, точность которой составляет 0,1 мм.
3. Оценка размеров печени осуществлялась путём измерения передне-задней величины правой доли печени.
4. Оценка размеров поджелудочной железы осуществлялась путём измерения переднезадней величины головки, тела и хвоста pancreas.
5. Оценка структуры паренхимы печени и поджелудочной железы определялась характером и равномерностью распределения отражённых эхосигналов, формированием характерных изображений нормальных анатомических образований органов.
6. Оценка эхогенности паренхимы печени и поджелудочной железы определялась степенью плотности отражённых эхосигналов, формирующих изображение, в каждом конкретном случае проводилось сравнение паренхимы обследуемого органа с эхогенностью близлежащей почки.
Состояние сосудистого рисунка печени оценивали путём измерения диаметра
1. основного ствола воротной вены, дополнительно проводилась визуальная оценка крупных и периферических сосудов печени.
2. Определение размеров желчного пузыря проводили на срезе с максимальным его изображением, измеряя продольный размер расстоянием от дна до шейки и поперечный размер – расстоянием между боковыми стенками, устанавливая маркеры электронной линейки строго перпендикулярно оси желчного пузыря.
3. Состояние полости желчного пузыря оценивали по эхогенности пузырной желчи, наличию или отсутствию внутриполостных образований.
У всех обследуемых детей с впервые выявленным сахарным диабетом пяти подгрупп УЗИ иллюстрирует увеличение в размерах pancreas; у всех детей определяется более выраженная гиперплазия хвостовой части ПЖ. (Табл. № 1, Рис.2). У детей 1-ой подгруппы (1-3 года) фиксируется увеличение головки, тела и хвоста ПЖ. У детей 2ой подгруппы (4-6 лет) обращает на себя внимание интенсивность увеличения головки и хвоста ПЖ.
Неоднородность ткани ПЖ определяется у детей с 4-х летнего возраста 12,5%, при чём скачок неоднородности ткани ПЖ замечен у детей с 10-12 лет 28,6% (4-ая подгруппа), 30% (5-ая под-группа), (Табл. № 4, Рис.3).
При УЗИ печени следует отметить интенсивную гепатомегалию у всех обследуемых, начиная с 1-ой подгруппы (1-3 года) и заканчивая 5-ой подгруппой (13-15 лет) см. Табл. № 3, Рис. 3.
Неоднородность ткани печени имеется у всех обследуемых. Обращает на себя внимание высокий процент неоднородности ткани печени у больных 1-ой подгруппы – 18,2 %, во 2-ой подгруппе – 12,3%, в 3-ей – 14,3%, скачок неоднородности ткани печени (как и поджелудочной железы) определяется в 4-ой подгруппе (10-12 лет) – 35,7%, в 5-ой подгруппе – 25,4% (Табл. № 4, Рис. 3).
Высокий процент уплотнённости стенки сосудов системы воротной вены наблюдается у всех обследуемых: 45,5 % в 1-ой подгруппе, 45,8 % во 2-ой, 35,7 % в 3-ей, 42,9 % в 4-ой, 65 % в 5-ой подгруппе (Табл. № 4, Рис.5).
Желчный пузырь неправильной формы определяется во всех подгруппах: 1-ая подгруппа у 9,1 % больных, 2-ая – у 16,7 %, 3-я – у 21,4 %, 4-я – у 28,6 %, 5-я – у 30 % больных.
Итак, при впервые выявленном СД у всех детей, независимо от пола и возраста, имеется значительное увеличение в размерах печени и ПЖ, наличие неоднородности ткани этих органов, заметен высокий процент уплотнённости стенки системы воротной вены. Следует отметить возрастной рост патологии желчного пузыря.
Выше приведенные данные при впервые выявленном сахарном диабете у детей иллюстрируют патологический процесс в pancreas, проходящий на фоне регрессивных изменений в hepar.
Увеличение в размерах ПЖ, изменения картины крови, биохимических показателей иллюстрируют наличие морфологических и функцио- нальных изменений в pancreas, начинающихся с воспалительного процесса, что ведёт к деструкции ткани ПЖ со становлением аутолиза ткани и следственным развитием инсулиновой недостаточности. Заключительную роль в развитии СД играет печень. Увеличение печени в размерах происходит в следствие экзогенных факторов (воспалительного процесса, повышенного всасы- вания продуктов гидролиза, липидов и моносаха- ридов, являющихся предшественниками триглицеридов) и эндогенных патогенетических факто- ров (усиление периферического липолиза, снижение утилизации жирных кислот гепатоцитами, блокада ферментов, участвующих в синтезе ЛПОНП и их секреции, дефицит белков в гепатоцитах, усиление гликогенолиза и др.). По мере снижения гликогена в печени и накопления жира печёночная клетка становится всё более уязви- мой и чувствительной к токсическим влияниям. Снижение секреции инсулина стимулирует липо- лиз, повышенное содержание глюкагона ускоряет образование кетоновых тел. Кетоз, повышенное образование кетоновых тел способствует нарушению жирового обмена с последующим избыточным накоплением жира в печени, развитием жирового гепатоза (стеатоза). Снижаются все функции печени и, конечно, углеводная – нарушается регуляция баланса между уровнем инсулина и глюкагона, т.е. наблюдается следственная инсулинорезистентность и глюкагонорезистентность. В завершение происходит развитие относительной, впоследствии – абсолютной инсулиновой недостаточности с клинической картиной сахарного диабета.
Базируясь на субъективных, объективных, данных лабораторного и инструментального обследования у детей с впервые выявленным сахарным диабетом, можно предположить, что в основе заболевания имеет место тотальное поражение pancreas (аутолиз) и параллельно идущие патологические изменения hepar (стеатоз).
Итак, сахарный диабет – это нейроиммунно-гландулярно-органное заболевание, характеризующееся нарушением всех видов обмена веществ на фоне тотального поражения pancreas (аутолиз) и жировой инфильтрации hepar (стеатоз) с развитием следственной глюкагонои инсулинорезистентности.
Для своевременной диагностики патологических изменений печени и поджелудочной железы у детей с впервые выявленным сахарным диабетом следует расширить объём биохимического обследования, добавив обязательное определение уровня триглицеридов, холестерина, ЛПНП, амилазы в стационарных условиях.
Взяв в основу многогранность патогенеза сахарного диабета, увидев причинноследственную связь в развитии заболевания, в лечении пациентов не стоит ограничиваться только назначением сахароснижающих препаратов (как правило, постоянной инсулинотерапии, даже в малых дозах).
Для предупреждения прогрессирования заболевания, раннего становления осложнений, лечение сахарного диабета должно быть комплексным. Фундаментом является строгий режим питания и диета (Стол № 9-5, ДП), полное исключение диетонеприемных продуктов (чипсы, кириешки), кофе, напитков (кока-кола и др.), устранение сусли, фрусли, ограниечение легко усвояемых углеводов (фруктоза, сорбит и др.). Щадящий вариант содержит физиологическую норму белка (1/3 белка животного происхождения) с ограничением жира и углеводов. Пища готовится на пару или даётся в отварном виде тёплой, жидкой или полужидкой консистенции. Режим питания – дробный до 6 раз в сутки.
Медикаментозное лечение должно состоять из сахароснижающих препаратов (инсулинотерапии, методика малых доз), инфузионной терапии, гепатопротекторов, ферментов, парентерального введения витаминов, антибактериальных и симптоматических препаратов.
С целью ранней профилактики сахарного диабета у детей, у новорождённых с наличием факторов риска сахарного диабета (предиабет), вакцинацию (гепатит, туберкулёз и др.) следует рассматривать как одно из существенных обстоятельств в патогенезе данного заболевания. Для уменьшения заболеваемости сахарным диабетом в группе детей высокого перинатального риска необходимо сместить вакцинацию с периода новорождённости на возраст от 1,5 до 2-х лет.
Литература
1. Абусуев, С. А., Хагиров, Д. Г. Пробл. эндокринол. 1996. № 5. С. 12–14.
2. Александрова, Н. И. Панкреатиты у детей / Н. И. Александрова, Г. М. Казакевич. Л.,1986.
3. Алексеев, Л. П. Сахарный диабет / Л. П.Алексеев [и др.]. 1998. № 1. С. 19–21.
4. Балаболкин, М. И. Эндокринология / М.И. Балаболкин. М.: «Универсум паблишинг», 1998.
5. Балаболкин, М. И. Роль окислительного стресса в патогенезе сосудистых осложнений диабета / М. И. Балаболкин, Е. М. Клебанова // Проблемы эндокрин. 2000. № 6. С. 29–34.
6. Балаболкин, М. И. Дифференциальная диагностика и лечение эндокринных заболеваний/ М. И. Балаболкин, Е. М. Клебанова, В. М. Креминская. М., 2002.
7. Баранов, В. Г. Сахарный диабет у детей/ В. Г. Баранов, А. С. Стройкова. Л.: «Медицина»,1980.
8. Белоусов, Ю. В. Заболевания поджелудочной железы у детей: панкреатит или панкреатопатия? – [Электронный ресурс.] // www. health – ua. com.
9. Белоусов, Ю. В. Педиатрическая гастроэнтерология: новейший справочник / Ю. В. Белоусов. М.: ЭКМО, 2006. 704 с.
10. Белякова, Н. А., Мазурова, В. И. Ожирение. СПб., 2003.
11. Благосклонная, Я. В, Шляхто, Е. В.,Бабенко, А. Ю. Эндокринология. СПб., 2007.
12. Буеверов, А. О. Жировая печень: причины и последствия / А. О. Буеверов // Журн.практик. врача. 2002. № 1. С. 36–38.
13. Бутрова, С. А., Дугоева, Ф. Х. Ожирение и метаболизм. 2004. № 1. С. 10–16.
14. Васильев, А. Ю. Ультразвуковая диагностика в детской практике / А. Ю. Васильев, Е. Б. Ольхова. М.: Изд. группа «ГЭОТАР-МЕДИА»,2007.
15. Диденко, В. А. Метаболический синдром Х: история вопроса и этиопатогенез / В. А. Диденко // Лабораторная медицина. 1999. № 2.
16. Завьялкин, В. А. Оптимизация диагностики и лечения острого панкреатита у детей: автореф. дис. канд. мед. наук / В. А. Завьялкин. СПб.,2006.
17. Завьялова, Н. Г. Ультразвуковая и радионуклеидная патология гепатобилиарной системы у детей дошкольного возраста: автореф. дис. … канд. мед. наук / Н. Г. Завьялова. Томск,1997.
18. Ивашкин, В. Т. Краткое руководство по гастроэнтерологии / В. Т. Ивашкин, Ф. И. Комаров, С. И. Рапопорт. М.: М-Вести, 2001. 430 с.
19. Касаткина, Э. П. Сахарный диабет у детей и подростков / Э. П. Касаткина. М., 1996.
20. Кудрякова, С. В. Проблемы эндокринологии / С. В. Кудрякова [и др.]. 1984. № 6. С. 3–6.
21. Клиническая эндокринология / под ред.Н. П. Старковой. Питер, 2002.
22. Лобанова М. В. Реогепатограмма у больных сахарным диабетом: тез. докл. 7-й Ресбулик. конф. эндокрин. Беларуси / М. В. Лобанова. Брест, 1979.
23. Лобанова, М. В. Изменения реогепатограммы у больных хроническим гепатитом в условиях изменения толерантности к глюкозе: тез. докл. научн. конф. гастроэнтерол. / М. В. Лобанова. Смоленск, 1979.
24. Лобанова, М. В. Гипофизарноглюкокортикоидный статус у больных сахарным диабетом с сопутствующей патологией печени: тез. докл. научн. конф. гастроэнтерол. / М. В. Лобанова, В. П. Лисун-Лобанова. Смоленск, 1980.
25. Лобанова, М. В. Гипогликемический синдром у больных с заболеванием печени и желчевыводящих путей: тез. докл. научн. конф. гастроэнтерол. / М. В. Лобанова. Смоленск, 1981.
26. Лобанова, М. В. Функциональная недостаточность надпочечников у больных сахарным диабетом с сопутствующей патологией печени: тез. докл. 2-го Всесоюзного съезда эндокринол. / М. В. Лобанова, В. П. Лисун-Лобанова, А. В. Байда. Ленинград, 1980.
27. Лобанова, М. В. Лечение больных сахарным диабетом с сопутствующей патологией печени: тез. докл. научн. конф. эндокринол., посвящ. 20-летию Литов. научн. общества эндокринол. / М. В. Лобанова. Каунас, 1982.
28. Лобанова, М. В. Анализ факторов риска для манифестации сахарного диабета у детей / М. В. Лобанова // Здравоохранение. 1998. № 9. С.5–7.
29. Лобанова, М. В. Первичная профилактика сахарного диабета 1 у детей и подростков / М. В. Лобанова // Медицинский журнал. 2007. №4. С. 125–127.
30. Лобанова, М. В. Метаболический синдром / М. В. Лобанова // Здравоохранение. 2008.№ 10. С. 39–43.
31. Мечников, И. И. Процесс «внутриклеточного пищеварения»: доклад на съезде врачей и естествоиспытателей / И. И. Мечников. Одесса,1883.
32. Полякова, С. И. Ранние проявления хронического панкреатита у детей: автореф. дис… канд. мед. наук / С. И. Полякова. М., 2003.
33. Рачинский, С. В., Сухановский, В. П., Гаврилов, А. А., Пономарева, Л. П., Барашнев, Ю. И., Таболин, В. А. Педиатр. М., 1998. № 1. С. 104–108.
34. Ревтович, М. Ю. Ингибиторы продукции цитокинов и перекисного окисления липидов в лечении острого панкреатита: автореф. дис. … канд. мед. наук / М. Ю. Ревтович. Минск, 2004.
35. Римарчук, Г. Принципы терапии хронического панкреатита у детей / Г. Римарчук, Н. И. Урсова, С. Л. Полякова // Российский педиатр. журнал. 2000. № 6. С. 27–31.
36. Старосельцева, Л. К. Содержание инсулина и глюкагона при различных формах сахарного диабета: тер. архив. / Л. К. Старосельцева [и др.]. 1980. № 8. С. 60.
37. Строгий, В. В. Инсулинорезистентность у детей и подростков как основа формирования метаболического синдрома / В. В. Строгий // Здравоохранение. 2006. № 4. С. 12–14.
38. Трофименко, Е. В. Некоторые эпидемиологические и иммунологические показатели инсулинозависимого сахарного диабета у детей: автореф. дис. … канд. мед. наук / Е. В. Трофименко. М., 1995.
39. Шабалов, Н. П. Детские болезни / Н. П.Шабалов. С.П., 2007.
40. Шевченко, О. П. Метаболический синдром / О. П. Шевченко, Е. А. Праскурничий, А. О. Шевченко. М., 2004.
41. Филлипович, Н. Е. Диагностика и лечение деструктивных форм острого панкреатита: автореф. дис. … д-ра мед. наук / Н. Е. Филлипович. Минск, 1986.
42. Яковенко, Э. П. Хронические заболевания печени, диагностика и лечение / Э. П. Яковенко, П. Я. Григорьев // Рус. мед. журнал. 2003. Т. 11. № 5. С. 291–296.
43. Яновская, Э. Ю. Педиатр. М., 2003. №1. С. 96–101.
44. Andress, R., Swerdloff, R., Rosefsky, T., Coleman, D. Manual Freedback Technique for the Control of Blood Glucose Concentration, in Automation in Analytical Chemistry / ed. A. Mediad. Skeggs LT, 1966. P. 486–491.
45. Angulo, P. Treatment of nonalcoholic fatty liver disease. Ann Hepatol 2002; 1:12–9. P, Keach JC, Batts KP, Lindord KD. Independent predictors of Liver fibrosis in patients with non-alcoholic steatohepatitis. Hepatology 1999;30:1356–62.
46. Burt, AD, Mutton, A, Day, CP. Diagnosis and interpretation of steatosis and steatohepatitis. Semin Diagn Pathol 1998; 15: 246–258.
47. Caro, J.F. J.Clin. endocrinol. metab.1991. Vol. 73. P. 691–695.
48. Classen, D.C., Classen, J.B. Infection Diseases in Clinical Practice. 1997 (6): 449–454.
49. Field, J. B. Influence of insulin on the hepatic up take and release of glucose and aminoacids / J.bB. Field. M: Hand book of Physiology. Sect. 7. V. I. Washington, 1972. P. 505.
50. Forbes, L. V., Scott, R. S., Brown, L. J., Darlow, B. A. Diabetes Care. 1995. Vol. 18, № 11. P.1428–1433.
51. De Fronzo, Ferrznini, E. Diabet.Care.1991. Vol. 4, № 3. P. 173–194.
52. Gale, E.A.M., Gillespie, K.M. Diabetologia.2001. Vol. 44, № 1. P. 3–15.
53. Hennes, M.M., Shrago, E., Kisseban, A. Obesity. 1980. Vol. 14. P. 831–841.
54. Kobberling, J., Bruggeboes, B. Diabetologia. 1980. Vol. 18, № 16. P. 459–462.
55. Kuntz, E. Fatty liver – a morphological and clinical review / E. Kuntz. Med Welt 1999; 50; 406–413.
56. Lefkowich, JH. Hepatobiliary patology / JH. Lefkowich. Curr Opin Gastroenterol 2003; 19:185–193.
57. Lumeng, L. Alcoholic liver disease / L. Lumeng, DW. Crabb. Curr Opin Gastroenterol 2000;16: 206–218.
58. Mirsky, I.A., Broh-Kahn, R.H. Inactivation of insulin by tissue extracts.I. The distribution and properties of the inactivating extracts (insulinase). Arch. Biohem., 1949. Vol. 20. P. 1.
59. Moseley, RH. Liver and biliary tract / RH.Moseley. Curr Opin Gastroenterol 2003;19:181–184.
60. Pessaure, D. Nonalcoholic steatosis and steatohepatitis. Mitochondrial dysfunction in steatohepatitis / D. Pessaure, A.M. Mansouri, B. Fromenty// Am J Physiol. 2002;282:193–199.
61. Reaven, G. M. Diabetes. 1988. Vol. 37. P. 1595–1607.
62. Sarles, H. Chronic inflammatory sclerosis of the pancreas – an autonomous pancreatic disease/ H. Sarles [et al.] // Am J Dig. Dis. 1961; 6: 688–699.
63. Williamson, R. F. The half life endogenous serum immunereactive insulin in men / R. F. Williamson, R. E. Gleason, J. S. Soeldner. Metabolism, 1968, Vol. 17. P. 1025.


Комментировать