Мозговой инсульт. Частота новых случаев ишемического инсульта среди лиц с сахарным диабетом: результаты проведенных исследований.
Мохорт Т.В.
Белорусского государственного медицинского университета
Мозговой инсульт – одно из наиболее частых проявлений при сахарном диабете (СД), эта патология развивается более чем у 700 000 пациентов в год. В США мозговой инсульт является 3-й причиной смерти и нетрудоспособности при СД. СД – наиболее важный фактор риска развития ишемических инсультов и тесно ассоциирован с повышением не только частоты заболеваемости, но и смертности[5].
По результатам многолетнего эпидемиологического исследования было установлено, что частота новых случаев ишемического инсульта среди лиц с сахарным диабетом достигает 62,3 на 1000 человек, в то время как в основной популяции она составляет 32,7 на 1000 человек в течение 12- летнего периода наблюдения [41, 4]. Относитель- ный риск развития инсульта выше у лиц с СД 2-го типа в 1,8–6 раз по сравнению с лицами без СД. В исследовании MRFIT риск смерти от инсульта среди пациентов с СД был в 2,8 раза выше по сравнению с пациентами без сахарного диабета, при этом риск смерти от ишемического инсульта был выше в 3,8 раза, от субарахноидального кро- воизлияния – в 1,1 раза и от внутримозгового кро- воизлияния – в 1,5 раза [39]. Вместе с тем, частота случаев геморрагического инсульта не отличается от таковой в общей популяции [29, 36]. СД является фактором риска развития нарушений мозгового кровообращения независимо от наличия других факторов риска (артериальная гипертензия, гипер- холестеринемия). У большинства (72–75%) больных сахарным диабетом отмечается не тромботи- ческий характер инсульта, что превышает стандартные показатели (среди населения в целом –60%). В развитии нетромботического инфаркта мозга при СД ведущие роли принадлежат хронической ишемии мозга и поражению симпатических вазомоторных нервов.
Существенную роль в развитии цереброваскулярных нарушений играет патология магистральных артерий головы: сонных и позвоночных артерий, которые при сахарном диабете часто поражаются атеросклерозом. Доказано, что СД и гипергликемия являются независимыми факторами риска развития системного атеросклероза с поражением сосу- дов различных локализаций, в том числе мозговых. При этом подавляющее большинство работ посвящены оценке атеросклеротического поражения коронарных артерий и артерий нижних конечностей, поражение которых сопровождается повышением в 2–4 раза риска смерти от инфаркта миокарда и ампутаций нижних конечностей [41]. Кроме того, для сахарного диабета характерно системное поражение сосудов микроциркуляторного русла (микроангиопатия), которое сопровождается развитием нарушений микроциркуляции в органемишени, т.е. мозге. Микроангиопатия мозговых сосудов усугубляет метаболические нарушения, развивающиеся при хронической ишемии мозга и повышает риск деменции [37].
Важность изучения влияния глюкозы и инсулина на толщину мышечного слоя артерий (индекс IMT) подтверждена работами, проводившимися в рамках международной программы IRAS [18].
Вторым проявлением цереброваскулярной патоло- гии при сахарном диабете является когнитивная дисфункция и синдром деменции [10, 12]. Согласно метаанализу К.V. Allen и соавт., в 8 из 9 пролонги- рованных исследований (длительностью от 5 до 12 лет) подтверждена связь СД с когнитивными нару- шениями и повышенным риском деменции [7]. При этом отмечается повышение риска развития болезни Альцгеймера при СД (см. рисунок).
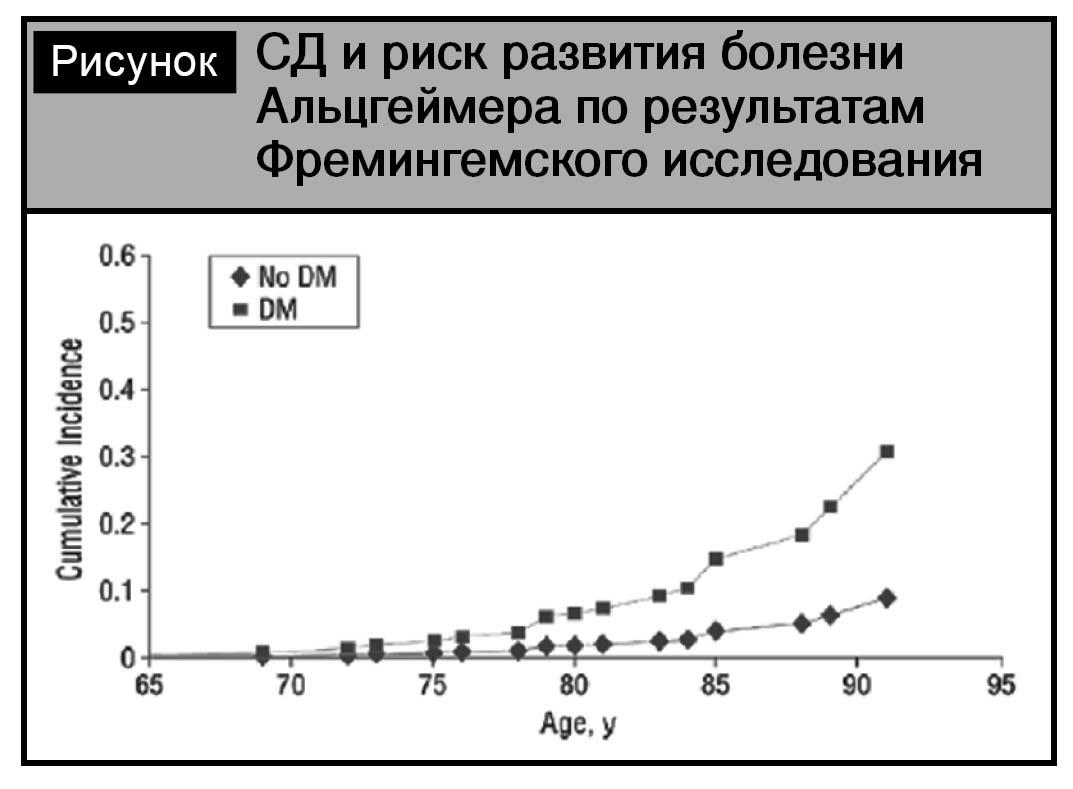
Доказано, что индекс гипогликемий нарастает с длительностью СД более 6 лет, при этом у пациентов выраженные гипогликемии ассоциированы с высоким риском деменции, а дополнительный рискдеменции между лицами без гипогликемических эпизодов и с зарегистрированными эпизодами со- ставил 2,39% в год [8]. Многочисленные публика- ции свидетельствуют, что при СД отмечаются снижение скорости психомоторных реакций, нарушение функции лобной доли, снижение вербальной памяти, комплексные моторные нарушения, визуальные задержки, снижение внимания и др. [13, 42].
Считается, что когнитивные и нейрональные нарушения при СД могут быть обусловлены прямым влиянием ятрогенных гипогликемий, другими метаболическими нарушениями (метаболический компонент деменции), нарушениями перфузии мозговой ткани (сосудистый компонент деменции), ассоциацией с депрессивными состояниями и другими факторами [10, 12].
Доказано, что острые и повторные гипогликемии снижают уровень гликогена в церебеллуме, коре и гипоталамусе на 50% по сравнению с нормальными значениями [19]. При этом после острой гипоглике- мии уровень гликогена возвращается к нормальным значениям через 6 ч, а при повторных гипогликемиях возращение уровня гликогена происходит через 24 ч, что связывают с нарушением контррегуляторных механизмов компенсации гиперинсули- немических гипогликемий [13, 43]. Есть мнение, что гипогликемия инициирует программированную гибель или повреждение нейронов за счет биоэнер-гетической дисфункции мозга [14, 42].
Патогенез деменции сложен, так как включает влияние различных факторов: нарушения макро- и микроциркуляции, изменения в ткани могза вследствие метаболических нарушений. Когнитивная дисфункция при СД связана с развитием глиоза и демиелинизации нервных волокон, повышением накопления миоинозитола с развитием атрофии в области гиппокампа и префронтальной зоны лоб- ной доли и сосудистыми нарушениями в белом веществе [24].
Развивающаяся при СД артериальная гипертензия, является причиной развития гипертонической энцефалопатии, вызывающей повреждение белого вещества головного мозга (лейкоареоз), сопровож- дающееся разобщением коры головного мозга и базальных ганглиев, т. е. нарушением кортикостри- атопаллидоталамических связей, играющих важ- ную роль в реализации когнитивных и двигатель- ных функций. Морфологические изменения в участ- ках лейкоареоза включают диффузную демиелини- зацию нервных волокон, пролиферацию и гипер- трофию астро-глии, отек олигодендроглиоцитов, очаги неполного некроза в белом веществе с обра- зованием мелких полостей, расширение перивен- трикулярных пространств, а также формирование спонгиоформной структуры белого вещества [33].
Основными факторами, инициирующими метабо- лические нарушения при СД, являются хроническая гипергликемия и выраженные гипогликемические эпизоды [11]. Хроническая гипергликемия определяет активцию инсулиннезависимых путей метабо- лизма глюкозы, в частности – полиолового шунта с накоплением глутатиона и активацией протеинки- назы С. Одновременно отмечается повышение внутриклеточной осмолярности с нарушением ра- боты K+/Na+ насоса; активируются реакции оксида- тивного стресса c накоплением свободно- радикальных продуктов и NO-зависимая вазодила- тация сосудов; процессы энзиматического и неэн- зиматического гликозилирования глюкозы; снижается продукция факторов роста (нейротрофилов), повышается адгезия лейкоцитов, активируются макрофаги и процессы свертывания крови. Перечисленные нарушения приводят к активации генов, определяющих повреждения нервной ткани и со- провождаются развитием глиоза, демиелинизацией и повышением накопления миоинозитола [15].
Выраженные гипогликемические эпизоды также оказывают негативное влияние на состояние мозга, так как нарушая питание и оксигенацию мозговой ткани вызывают селективные нейрональные некро- зы; сопровождаясь повышением уровня кальция, оказывают токсическое внутриклеточное действие и др. [35].
Тяжесть повреждения мозга при СД определяется cтепенью и длительностью снижения мозгового кровотока и нарушениями метаболических реакций мозга [5, 13, 30]. При снижении кровотока сначала происходит торможение белкового синтеза, затем отмечается активация анаэробного гликолиза, увеличение концентрации лактата с развитием лактат- ацидоза и тканевого цитотоксического отека. Следующая стадия характеризуется снижением синте- за АТФ, дисфункцией каналов активного ионного транспорта, дестабилизацией клеточных мембран, выбросом возбуждающего нейротрансмиттера глу- тамата, и при уменьшении кровотока до 20% от нормы развивается аноксическая деполяризация мембран, необратимое поражение клеток. В центре ишемического очага в течение нескольких минут наступает некроз, а вокруг «ядра» инфаркта распо- лагается ишемическая полутень, где изменения возможно обратимы. Именно зона ишемии является основной точкой приложения метаболических средств при остром или хроническом ишемическом поражении мозга. Основными механизмами по- вреждения мозга при ишемии являются глутама- тергическая эксайтотоксичность и оксидантный стресс [32].
При инсульте отмечается повышение содержания глутамата в нейронах, в цереброспинальной жид- кости и в крови, что вызывает приток ионов кальция (Са2+) внутрь клетки через NMDA-рецепторы (N- метил-D-аспартатного рецептора). Кроме того, глу- тамат участвует в образовании оксида азота (NO) и активации протеинкиназы С, протеаз, эндонуклеаз, фосфолипаз и протеинолиза, а также активации перекисного окисления липидов клеточных мем- бран [30, 32].
Другой важнейший патогенетический механизм апоптоза нейронов в условиях ишемии — оксида- тивный стресс, характеризующийся образованием избытка свободных радикалов, способных по- вреждать белки, жиры и ДНК. Оксидантный стресс развивается при нарушении соотношения между продукцией реактивных форм кислорода и антиок- сидантной защитой организма (супероксиддисмутаза, каталаза, витамины Е, С). Нейроны, особенно клетки стриатума, гиппокампа и коры головного мозга, восприимчивы к оксидативному поврежде- нию, что определяет возможность дополнительных влияний и обусловлено большим спектром липи- дов, участвующих в свободнорадикальных реакци- ях. Токсические проявления, развивающиеся в нервной ткани, результируются в развитие локаль- ного воспаления и активацию апоптоза.
Клинические проявления цереброваскулярной патологии при СД весьма разнообразны.
Лейкоареоз, характеризующий гипертонические и постгипогликемические поражения мозга, может быть бессимптомным или проявляться сочетанием когнитивных расстройств (без очаговых нарушений высших корковых функций), прогрессирующих до синдрома деменции, и различных неврологических нарушений (обмороки, судорожный, подкорковый, псевдобульбарный и мозжечковый синдромы) [1]. При этом пациенты предъявляют ряд жалоб на головные боли, головокружение, изменения настрое- ния, чаще с депрессивными симптомами.
Частота выявления цереброваскулярной патологии определяет необходимость использования различных лекарственных средств с учетом понимания различных патогенетических подходов к достижению нейропротекторного эффекта.
Одним из патогенетических подходов для достиже- ния этой цели является метаболическая терапия, направленная на улучшение пластичности здоро- вой ткани, усиление регенераторно-репаративных процессов, активацию образования полисинапти- ческих связей, увеличение количества рецепторов. Среди препаратов из группы активаторов метабо- лизма используются различные сывороточные ге- модериваты (CГД) – депротеинезированные дери- ваты из крови здоровых молочных телят, содержа- щие широкий спектр низкомолекулярных компонен- тов клеточной массы и сыворотки крови с молеку- лярной массой 5000 Д (гликолипиды, нуклеозиды и нуклеотиды, олигопептиды и аминокислоты) [2, 17,21, 26].
Проведенные экспериментальные исследования подтвердили, что депротеинезированные дериваты из крови здоровых молочных телят снижают глута- матергическую цитотоксичность нейронов, что поз- воляет объяснить один из механизмов эффекта воздействия на мозг. Кроме того, CГД обладают выраженным антиоксидантным эффектом, ингибируя проявления активции оксидативного стресса и оказывают влияние на процессы эксайтотоксично- сти – избыточной активации аминокислотных ре- цепторов нейронов [27]. Доказано, что сывороточ- ные гемодериваты являются мощными антиокси- дантами, так как они обладают высокой суперок- сиддисмутазной активностью и обеспечивают сни- жение уровней свободных радикалов [16]. В то же время сывороточные гемодериватыстимулируют утилизацию головным мозгом глюкозы в условиях гипоксии за счет усиления ее транспорта и накоп- ления в клетках, а также увеличивают поглощение кислорода тканями и оказывают инсулиноподобное действие, повышая толерантность организма к глюкозе.
Механизм действия сывороточных гемодериватов обеспечивает возможность оптимизации пути энер- гетического обеспечения, в результате чего увеличивается синтез макроэргических соединений и улучшаются метаболиче-ские процессы в пораженных тканях мозга и поддерживается белково- синтетическая функция нейронов [27].
Нейрогенные эффекты сывороточных гемодерива- тов используются с 1965 г., когда были доказаны положительные влияния на течение дисциркуля- торной энцефалопатии. В дальнейшем были про- ведены исследования, подтвердившие эффектив- ность этой группы препаратов в лечении когнитив- ных и других нарушений у пациентов с хронической ишемией мозга, инсультом, являющихся частой па- тологией, регистрируемой при СД [25, 38]. Доказа- но, что под влиянием сывороточных гемодеривато- вулучшаются мыслительные процессы, концентра- ция внимания, память, уменьшается интенсивность головной боли ишемически-гипоксического харак- тера [25, 40]. Позитивный эффект CГД «Актовегин» подтвержден в многоцентровом рандомизирован- ном исследовании при оценке состояния диабети- ческой дистальной нейропатии, в котором доказано улучшение течения и уменьшение проявлений нейропатии при отсутствии негативного влияния препарата на углеводный обмен [44].
Актовегин содержит ряд основных микроэлементов – натрий, калий, кальций, фосфор, магний. Магний является каталитическим центром всех известных сегодня нейропептидов головного мозга и имеет статус нейроседативного иона, что имеет принципиальное значение для обеспечения нейропротек- тивного эффекта.
Сывороточные гемодериваты эффективно используются в комплексной терапии мозгового инсульта (ишемического и геморрагического). При этом наряду со значимым уменьшением субъективных и объективных симптомов отмечается достоверное улучшение мозгового кровотока.
В результате лечения ишемического инсульта у 82% больных отмечено субъективное улучшение состояния, а у 61% – объективное уменьшение неврологической симптоматики [9]. При ишемических формах инсультов целесообразно назначать метаболическую терапию как можно раньше, так как в это время происходит активация перекисного окисления липидов при ишемии [3, 34]. Однако учи- тывая закономерности эксайтотоксичности, имеют- ся рекомендации начинать введение нейропротек- торов после 10–30-минутной реперфузии – по до- стижении максимального уровня продуктов пери- кисного окисления липидов в ишемическом очаге.
СГД используются при геморрагических инсультах, обеспечивая снижение медленно-волновой актив- ности на ЭЭГ в области очага кровоизлияния, сни- жение риска возникновения сосудистого спазма, развития стойкого неврологического дефицита и последующей инвалидизации [28]. Достигнутые эффекты связывают с оксигенацией окружающих очаг тканей, снижением уровня лактата и нормали- зацией электрической активности нейронов коры. Поскольку сывороточные гемодериваты повышают потребление кислорода клетками, авторы не реко- мендуют использовать его высокие дозы в острей- шем периоде геморрагического инсульта, в сочета- нии с угнетением сознания. Лучший эффект ис- пользования СДГ при геморрагическом инсульте отмечается на 4–7-е сутки после внутримозгового кровоизлияния, когда уменьшаются явления отека мозга.
В восстановительном периоде инсульта сывороточные гемодериваты приводят к улучшению пси- хических и когнитивных нарушений, обеспечивая улучшение речевых и двигательных функций, регресс психических симптомов беспокойства и воз- буждения, нарушения поведения [20, 21].
Перечисленные эффекты позволяют прогнозиро- вать эффективность сывороточные гемодериваты при дисциркуляторной энцефалопатии в отношении когнитивных (запоминание новой информации и воспроизведение уже имеющейся), двигательных и психических нарушений [23, 26, 31]. Отмечено субъективно значительное улучшение общего состояния у 44% пациентов, у 47% больных отмечен значимый регресс двигательных нарушений. По- вышение интереса к пище, прогулкам, общению с окружающими отмечены у 66,7%; снижение тре- вожности, повышенной возбудимости – у 64,3%, улучшение качества сна – у 54,5%. Аффективные нарушения уменьшились у 67,9% пациентов, ипо- хондрия – у 60,6%.
Таким образом, сывороточные гемодериваты – универсальные препараты, которые могут быть использованы при сосудистых и метаболических по- ражениях головного мозга. Обладая антиоксидант- ными и другими метаболическими эффектами, СГД активизируют нейрональный метаболизм и микро- циркуляцию, оказывают позитивное влияние на те- чение дистальной формы нейропатии и являются безопасными и не оказывают отрицательного вли- яния на метаболизм углеводов [22]. В то же время необходимо проведение исследований по оценке церебропротективного эффекта у пациентов с СД, осложненным различными циркуляторными нару- шениями и при развитии когнитивных нарушений и деменции, обусловленных выраженными симпто- матическими гипогликемиями.
ЛИТЕРАТУРА
1. Левин О.С., Дамулин Н.В. Диффузные измене- ния белого вещества (лейкоapeoз) и проблема сосудистой деменции // Достижения в нейроге- риатрии: сборник научных трудов / Под ред. Н.Н. Яхно и И.В. Дамулина. – М., 1995. – 293 с.
2. Шмырев В.И., Остроумова О.Д., Боброва Т.А. // Рус. мед. журн. – 2003. – Т. 11, № 4. – С. 216–220.
3. Янсен В., Брукнер Г.В. // Рус. мед. журн. –2002. – Т. 10, № 12–13. – С. 543–546.
4. Abbott R.D., Donahue R.P., MacMahon S.W. et al.// JAMA. – 1987. – Vol. 257. – P. 949–952.
5. Air E.L., Kissela B.M. // Diabetes Care. – Vol. 30. –P. 3131–3140.
6. Akomolafe A., Beiser A., Meigs J.B. et al. // Arch.Neurol. – 2006. – Vol. 63. – P. 1551–1555.
7. Allen K.V., Frier B.M., Strachan M.W. // Eur. J.Pharmacol. – 2004. – Vol. 490. – P. 169–175.
8. Amiel S.A., Dixon T., Mann R., Jameson K. // Diabet. Med. – 2008. – Vol. 25, N 3. – P. 245–254.
9. Araki G. // Kiso to Rinsho. – 1974. – Vol. 8. –P. 4208–4214.
10. Biessels G.J., Staekenborg S., Brunner E. et al. // Lancet Neurol. – 2006. – Vol. 5. – P. 64–74.
11. Boyle P.J. // Diabetologia. – 1997. – Vol. 40, Suppl 2. – S69–S74.
12. Christopher T.K., Elizabeth R. // Endocr. Rev. –2008. – Vol. 29, N 4. – P. 494–511.
13. Cryer P.E. // J. Clin. Invest. – 2007. – Vol. 117, N 4. – P. 868–870.
14. Cryer P.E. // Diabetologia. – 2002. – Vol. 45. –P. 937–948.
15. Cox D.J., Kovatchev B.P., Gonder-Frederick L.A.
et al. // Diabet. Care. – 2005. – Vol. 28. – P. 71–77.
16. De Groot B.M., Machicao F. // Res. Comm. Chem.Path. Pharm. – 1990. – Vol. 68. – P. 125–128.
17. Derev’yannykh E.A., Bel’skaya G.N., Knoll E.A. et al. // Neurosci. Behav. Physiol. – 2008. – Vol. 38,N 8. – P. 873–875.
18. Festa A., D’Agostino R.J., Mykkanen L. et al. // Arterioscler. Thromb. Vasc. Biol. – 1999. – Vol.19. – P. 562–568.
19. Gelling R.W., Morton G.J., Morrison C.D. et al. // Cell Metabolism. – 2006. – Vol. 3, N 1. – P. 67–73.
20. Herrmann W.M., Bohn-Olszewsky W.J., Kuntz G.// Z Geriatrie. – 1992. – Vol. 5. – P. 46–55.
21. Herrschaft H., Kunze U., Gleim F. // Med. Welt. –1977. – Bd. 28. – S. 339–345.
22. Jacob S., Dietze G.J., Machicao F. et al. // Arzneimittelforschung. – 1996. – Bd. 46. – P. 269–272.
23. Jacobson A.M., Musen G., Ryan C.M. et al. // NEJM. – 2007. – Vol. 356. – P. 1842–1852.
24. Kane W.C., Aronson S.M. // Am. J. Pathol. –1968. – Vol. 52. – P. 71–75.
25. Kanowski S., Kinzler E., Lehmann E. et al. // Pharmacopsychiatry. – 1995. – Vol. 28. – P. 125–133.
26. Klein K., Siedek H. // Med. Welt. – 1965. – Vol.13. – P. 647–650.
27. Kume T., Asai N., Nishikava H. et al. // Proc. Natl.Acad. Sci. USA. – 2002. – Vol. 99. – P. 3288–3293.
28. Lencher H., Logar C., Grieshofer P. // New Trends in Clin. Neuropharmacol. – 1991. – Vol. 5. – P.147–152.
29. Megherbi S.E., Milan C., Minier D. et al. //Stroke. – 2003. – Vol. 34. – P. 688–694.
30. Miyamoto E. // J. Pharmacol. Sci. – 2006. – Vol.100. – P. 433–442.
31. Nagatsuka K., Tsuda Y., Takano T. et al. // Geriat.Med. – 2002. – Vol. 26. – P. 1202–1215.
32. Parsons C.G., Danysz W., Quack G. // Drug NewsPerspect. – 1998. – Vol. 11. – P. 523–569.
33. Peress N.S., Kane W.C. // Prog. Brain Res. –1973. – Vol. 40. – P. 473–483.
34. Reeves M.J., Vaidya R.S., Fonarow G.C. et al. // Stroke. – 2010. – Vol. 41, N 5. – P. e409–e417.
35. Ryan C.M., Williams T.M., Finegold D.N., Orchard T . J. // Diabetologia. – 1993. – Vol. 36, N 4. – P. 329–334.
36. Sander D., Sander K., Poppert H. // Brit. Journ.Diabetes & Vasc. Disease. – 2008. – Vol. 8, N 5. –P. 222–229.
37. Schmidt R., Schmidt H., Fazekas F. // J. Neurol. –2000. – Vol. 247. – P. 81–87.
38. Semlitsch H.V., Anderer P., Saletu B., Hochmayer I. // Neuropsychobiol. – 1990/91. –Vol. 24. – P. 49–56.
39. Stamler J., Vaccaro O., Neaton J.D. et al. // Diabetes Care. – 1993. – Vol. 16. – P. 434–444.
40. Talypov A.E., Ioffe I.S., Miatchin M.I.,Kuksova N .S. // Zh. Nevrol. Psikhiatr. Im. S.S. Korsakova. – 2008. – Vol. 108, N 8. – P. 20–23.
41. Tuttolomondo A., Pinto D., Di Raimondo A., Licata G. // Diabetes Care. – 2007. – Vol. 30, N 11. – P. e114–e124.
42. Warren R.E., Frier B.M. // Diabetes Obes.Metab. – 2005. – Vol. 7, N 5. – P. 493–503.
43. Wright R.J., Frier B.M. // Diabetes Metab. Res.Rev. – 2008. – Vol. 24, N 5. – P. 353–363.
44. Ziegler D., Movsesyan L., Mankovsky B. et al. // Diabetes Care. – 2009. – Date of access:doi:10.2337/dc09-0545.
Данная статья взята из журнала «Медицинские новости», № 6, 2011.






Комментировать