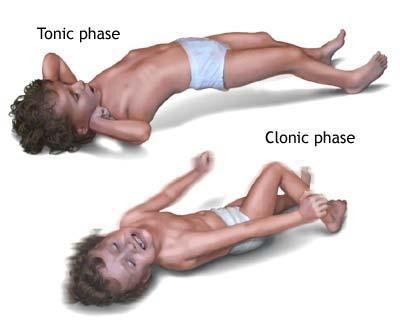
Эпилепсия в детском возрасте: разделение по возрастным группам – дошкольники, школьники и подростки. Описание видов эпилепсий, согласно возрастным группам.
В. М. Студеникин , доктор медицинских наук, профессор, член-корреспондент РАЕ, ФГБУ «НЦЗД» РАМН, Москва
Эпилепсии у детей дошкольного возраста (4–6 лет)
Для детей дошкольного возраста характерен дебют идиопатической парциальной эпилепсии с лобными пароксизмами, доброкачественной затылочной эпилепсии с ранним дебютом (синдром Панайиотопулоса), а также синдрома Ландау–Клеффнера.
Идиопатическая парциальная эпилепсия с лобными пароксизмами
Болезнь впервые описали A. Beaumanoir и A. Nahory (1983). Возраст пациентов к моменту дебюта эпилепсии составляет 2–8 лет. На этот тип эпилепсии приходятся около 11% случаев всех идиопатических фокальных эпилепсий. Болезнь манифестирует в виде нескольких типов приступов: дневных (сложные парциальные, моторные автоматизмы, иногда абсансоподобные) и ночных (гемифациальные моторные приступы, версивные, иногда с «посткризисным» дефицитом и/или вторично-генерализованными припадками). Частота приступов варьирует от 1 эпизода в месяц до 1 припадка за несколько недель (продолжительность активного периода болезни составляет от 1 года до 6 лет). Данные ЭЭГ достаточно гетерогенны и не имеют единого специфического паттерна (у части пациентов ЭЭГ-изменения сопоставимы с таковыми при доброкачественной детской эпилепсии с центротемпоральными пиками; у других имеется только очаговая медленная активность; в иктальном периоде регистрируются перемежающиеся лобные разряды). Прогноз болезни довольно благоприятен (спонтанная ремиссия). Транзиторное снижение когнитивных функций (кратковременной памяти, оперативных функций и др.) отмечается в течение активного периода заболевания; затем они постепенно восстанавливаются [3–6].
Доброкачественная затылочная эпилепсия с ранним дебютом (Панайиотопулоса синдром)
Одна из доброкачественных затылочных эпилепсий детского возраста. Болезнь дебютирует в возрасте от 1 года до 14 лет (пик встречаемости в 4–5-летнем возрасте); встречается примерно в 2 раза чаще, чем вариант Гасто. Характерны автономные ночные приступы; вследствие вегетативной симптоматики и тошноты припадки плохо распознаются. На ранних стадиях болезни отмечаются девиация глаз и поведенческие расстройства (в 50% случаев приступы могут приобретать судорожный характер). Продолжительность приступов составляет 5–10 мин; у 35–50% пациентов они переходят в автономный фокальный эпилептический статус (иногда с вторичной генерализацией). При ЭЭГ регистрируются пики или пароксизмальные разряды. У двух третей пациентов отмечается по меньшей мере одно ЭЭГ-исследование с признаками затылочных пароксизмов (чаще всего — затылочных пиков); у оставшейся трети больных отмечаются только внезатылочные пики или короткие генерализованные разряды. Примерно в 33% случаев у детей с синдромом Панайиотопулоса на ЭЭГ регистрируются мультифокальные пики в двух и более церебральных областях (единичные пиковые очаги считаются редкостью). Прогноз при синдроме Панайиотопулоса в отношении длительной ремиссии и когнитивных функций сравнительно благоприятен. Лечение доброкачественной затылочной эпилепсии с ранним дебютом преимущественно нацелено на контроль приступов в острой фазе (диазепам) [3–6].
Синдром Ландау–Клеффнера (приобретенная эпилептическая афазия)
Подавляющее число случаев заболевания приходится на возраст 4–5 лет, хотя болезнь (приобретенная афазия и эпилептиформные разряды в височных/теменных областях мозга) может дебютировать и раньше — на втором или третьем году жизни. Причина этого сравнительно редкого состояния неизвестна. Синдром Ландау–Клеффнера характеризуется утратой речевых навыков (афазия импрессивная и/или экспрессивная). Указанные речевые нарушения, а также слуховая агнозия появляются у детей, не имевших ранее отклонений со стороны психомоторного и речевого развития. Сопутствующие эпилептические припадки (фокальные или генерализованные тонико-клонические судороги, атипичные абсансы, парциальные сложные, редко — миоклонические) регистируются у 70% пациентов. У части детей отмечаются нарушения поведения. В остальном в неврологическом статусе у пациентов выраженные нарушения обычно отсутствуют. Специфический ЭЭГ-паттерн для синдрома Ландау–Клеффнера не характерен; регистрируются эпилептиформные разряды в виде повторных пиков, острых волн и пик-волновой активности в височных и теменно-затылочных областях головного мозга. Эпилептиформные изменения в состоянии сна усиливаются или отмечаются исключительно во время сна. Хотя прогноз синдрома Ландау–Клеффнера сравнительно благоприятен, дебют болезни в возрасте до 2 лет всегда сопряжен с неблагоприятным исходом по обретению и/или восстановлению навыков речевого общения [3–5, 9].
Эпилепсии у детей школьного возраста и подростков
По достижении школьного возраста дети и подростки подвергаются риску подверженности целой группы возрастзависимых эпилепсий, среди которых наиболее актуальны детская абсансная, доброкачественная с центротемпоральными пиками, доброкачественная затылочная (вариант Гасто), ювенильная абсансная, ювенильная миоклоническая (синдром Янца) и целый ряд других форм болезни.
Детская абсансная эпилепсия (пикнолепсия)
Пик дебютов болезни приходится на ранний школьный возраст (около 7 лет), хотя детская абсансная эпилепсия может дебютировать с 2 до 12 лет. До наступления 3-летнего возраста дебютирует редко. Относится к идиопатическим формам болезни; все случаи считаются генетически детерминированными (аутосомно-доминантный тип наследования с неполной пенетрантностью). Детская абсансная эпилепсия характеризуется частыми повторными приступами абсансов (до нескольких сотен за сутки). При этом абсансы представляют собой единственный или ведущий тип эпилептических приступов; вероятность появления генерализованных тонико-клонических судорог значительно ниже, чем при юношеской (ювенильной) абсансной эпилепсии. Диагностика детской абсансной эпилепсии осуществляется по клиническим проявлениям и данным ЭЭГ (типичный ЭЭГ-паттерн — вспышки генерализованной высокоамплитудной пик-волновой активности с частотой 3 за 1 сек, внезапно возникающие и плавно прекращающиеся). Прогноз при детской абсансной эпилепсии сравнительно благоприятен [3, 5, 6].
Доброкачественная эпилепсия детского возраста с центротемпоральными пиками (роландическая эпилепсия)
Описана P. Nayrac и M. Beaussart (1958). В возрастной группе 0–15 лет встречается с частотой 5–21 на 100 тыс. (8–23% от всех случаев эпилепсии), являясь самой распространенной формой идиопатической эпилепсии у детей. Возраст детей к моменту дебюта роландической эпилепсии варьирует от 3 до 14 лет (пик — 5–8 лет). Случаи болезни у пациентов в возрасте до 2 лет исключительно редки. Болезнь проявляется в виде ночных тонико-клонических приступов с парциальным (фокальным) началом, а также дневных простых парциальных (исходящих из нижних корковых отделов — области центральной роландической борозды). Частота приступов обычно невысока. Для роландической эпилепсии характерна специфическая соматосенсорная аура (патологические ощущения в щечно-ротовой области), а также гиперсаливация, остановка речи, тонико-клонические или тонические судороги фациальной мускулатуры. Сознание в момент приступа пациентом не утрачивается. Для ЭЭГ-данных характерно наличие пик-волновых комплексов, локализованных в центрально-височных отделах. В интериктальном периоде у пациентов при ЭЭГ регистрируются специфические комплексы в виде высокоамплитудных 2-фазных пиков, сопровождаемых медленной волной. Роландические пики локализуются в одном или обоих полушариях (изолированно или группами: в средневисочных — Т3, Т4, или центральных — С3, С4 — областях). К моменту дебюта болезни психомоторное развитие детей является нормальным. Впоследствии болезнь характеризуется практически полным отсутствием неврологического и интеллектуального дефицита. У многих пациентов в подростковом возрасте наступает ремиссия; у незначительной части детей отмечаются нарушения когнитивных функций (вербальная память), а также различные речевые расстройства и снижение успеваемости (в школе) [3–6, 9].
Доброкачественная затылочная эпилепсия с поздним дебютом (вариант Гасто)
Фокальная форма идиопатической эпилепсии детского возраста (синдром Gastaut) с более поздним началом, чем при синдроме Панайиотопулоса. Болезнь дебютирует в возрасте 3–15 лет, но пик приходится примерно на 8 лет. Характерны короткие по продолжительности приступы со зрительными нарушениями (простые и сложные зрительные галлюцинации), полной/частичной утратой зрения и иллюзиями, девиация глаз, вслед за которыми отмечаются клонические судороги, вовлекающие одну сторону тела. До 50% пациентов испытывают по окончании приступа мигренозные или мигренеподобные цефалгии. ЭЭГ-данные напоминают таковые при синдроме Панайиотопулоса: в интериктальном периоде регистрируется нормальная основная активность фоновой записи в сочетании с эпилептиформными одно- или двухсторонними разрядами в затылочных отведениях в виде пик-волновых комплексов (высокоамплитудные двухфазные пики с основной негативной фазой и следующей за ней непродолжительной позитивной фазой) в сочетании с негативной медленно-волновой активностью. При открытии пациентом глаз эпилептиформная активность исчезает, но снова возобновляется через 1–20 сек после закрытия глаз. Прогноз при доброкачественной затылочной эпилепсии с поздним дебютом (вариант Гасто) сравнительно благоприятен, но, в связи с возможной фармакорезистентностью болезни, неоднозначен [3–5, 9].
Ювенильная (юношеская) абсансная эпилепсия
Юношеский вариант абсансной эпилепсии относится к идиопатическим генерализованным формам болезни. В отличие от детской абсансной эпилепсии, болезнь обычно дебютирует в пубертатном, пре- или постпубертатном возрасте (9–21 год, чаще 12–13 лет). Болезнь проявляется типичными абсансами, миоклонусами или генерализованными тонико-клоническими судорогами. Вероятность дебюта в виде генерализованных тонико-клонических приступов при ювенильной абсансной эпилепсии несколько выше (41% случаев), чем при детской абсансной. Характерный для этого вида эпилепсии ЭЭГ-паттерн имеет вид пик-волновой активности с частотой 3 Гц — симметричный и билатерально-синхронизированный. Полипик-волновая активность, встречающаяся у части пациентов, должна настораживать в плане трансформации болезни в юношескую миоклонус-эпилепсию. Прогноз относительно благоприятен — высока вероятность наступления ремиссии в позднем подростковом возрасте [3–6, 9].
Ювенильная (юношеская) миоклонус-эпилепсия (синдром Янца)
Болезнь описана D. Janz и W. Christian (1957) в качестве одного из подтипов идиопатической генерализованной эпилепсии (другое название: «импульсивный petit mal»). Обычно дебютирует в возрасте 8–26 лет (чаще в 12–18 лет). Отличительным признаком болезни являются миоклонические припадки. Характерны изолированные миоклонические подергивания в верхних конечностях, особенно вскоре после пробуждения. У большинства детей отмечаются генерализованные тонико-клонические приступы, а примерно у трети пациентов имеются абсансы. Припадки часто провоцируются депривацией сна. Миоклонические приступы сопровождаются короткими вспышками генерализованных пик-волновых или полипик-волновых комплексов при проведении ЭЭГ [3–5, 9].
Височная семейная эпилепсия
Этот генетически гетерогенный синдром характеризуется сравнительно доброкачественными простыми или сложными парциальными (фокальными) приступами с выраженной психической или автономной аурой. Болезнь обычно дебютирует на втором (примерно 11 лет) или в начале третьего десятилетия жизни (чаще у совершеннолетних индивидов). Обычно возникает на фоне нормального развития ЦНС. При МРТ головного мозга не обнаруживается каких-либо патологических структурных изменений в области гиппокампа или височных долях. Данные ЭЭГ позволяют зарегистрировать эпилептиформную активность в срединных и/или латеральных областях височных долей. Припадки при семейной височной эпилепсии чаще легко поддаются медикаментозному контролю традиционными антиэпилептическими препаратами [3–6, 9].
Мезиально-височная эпилепсия
Чаще дебютирует у подростков и проявляется лимбическими приступами. В типичных случаях у больных, в анамнезе у которых отмечались фебрильные судороги, после свободного от приступов интервала возникают височные припадки, которые в первое время хорошо поддаются медикаментозному контролю. Впоследствии, в подростковом возрасте или по достижении совершеннолетия, отмечаются рецидивы болезни. При МРТ головного мозга у пациентов может обнаруживаться склероз гиппокампа, что считается ключевым признаком этой формы эпилептического синдрома. Все лимбические припадки в большей или меньшей мере рефрактерны к фармакотерапии [3, 5].
Cемейная мезиально-височная эпилепсия
Этот генетически детерминированный гетерогенный эпилептический синдром описали P. Hedera и соавт. (2007). Болезнь дебютирует в различном возрасте, но чаще всего на втором десятилетии жизни. В отличие от описанной выше мезиально-темпоральной эпилепсии, у детей в анамнезе обычно отсутствуют указания на фебрильные судороги. В большинстве случаев у пациентов отмечаются простые фокальные приступы с появлениям déjà vu, периодически ассоциированные с оглушенностью или тошнотой, в других случаях — сложные парциальные припадки с изменениями сознания и замираниями; реже имеют место вторично-генерализованные приступы. У части больных при МРТ отсутствуют признаки склероза гиппокампа или иных аномалий церебральных структур. Патологические изменения данных ЭЭГ примерно у половины пациентов отсутствуют. Менее половины случаев семейной мезиально-височной эпилепсии требуют проведения терапии антиэпилептическими препаратами [3, 5, 6].
Парциальная (фокальная) аутосомно-доминантная эпилепсия со слуховыми стимулами
Эта форма болезни фактически является одним из подтипов латеральной височной эпилепсии; она известна также под названием «телефонная эпилепсия». Болезнь дебютирует в возрасте 8–19 лет (чаще всего — на втором десятилетии жизни). Парциальная аутосомно-доминантная эпилепсия со слуховыми стимулами характеризуется слуховыми нарушениями (ощущение больным недифференцированных звуков и шумов), слуховыми галлюцинациями (изменения восприятия громкости и/или высоты звуков, голоса «из прошлого», необычное пение и т. д.). Помимо слуховых нарушений и галлюцинаций для этой формы болезни свойственны различные вегетативные расстройства, патологическая двигательная активность, а также многочисленные сенсорные и психические нарушения различной выраженности. В межприступном периоде при ЭЭГ у пациентов может наблюдаться парокcизмальная активность в височных или затылочных отведениях (или полностью отсутствовать) [3, 5, 9].
Эпилепсия с приступами grand mal при пробуждении
У детей дебют приступов при этой идиопатической генерализованной эпилепсии преимущественно происходит на втором десятилетии жизни. По проявлениям болезнь несколько напоминает ювенильную миоклонус-эпилепсию Янца. Развитие тонико-клонических приступов происходит исключительно или преимущественно после пробуждения (> 90% случаев) или вечером в периоде релаксации. Припадки вызываются депривацией сна. В отличие от миоклонус-эпилепсии Янца, миоклонии и абсансные приступы у пациентов с описываемой формой эпилепсии наблюдаются редко. ЭЭГ позволяет зарегистрировать генерализованную пик-волновую активность и признаки фотосенситивности (последние присутствуют не всегда) [3–6, 9].
Болезнь Унферрихта–Лундборга (балтийская или финская миоклонус-эпилепсия)
Эта редкая форма эпилепсии дебютирует у детей в возрасте от 6 до 13 лет (чаще примерно в 10-летнем возрасте). По своим проявлениям напоминает синдром Рамсэя Ханта. Первыми симптомами являются судорожные приступы. Миоклонии присоединяются по прошествии 1–5 лет; они отмечаются преимущественно в проксимальных мышцах конечностей, носят двухсторонний симметричный характер, но асинхронны. Миоклонии индуцируются фотосенсибилизацией. Выраженность миоклоний постепенно нарастает. Впоследствии происходит прогрессирующее снижение интеллекта (до степени деменции). На поздних этапах болезни у пациентов появляются признаки мозжечковой атаксии [3, 5, 6].
Ювенильный нейрональный цероидный липофусциноз тип III
Эта болезнь известна также под названием «прогрессирующая эпилепсия с умственной отсталостью» или «северная эпилепсия». Является представителем нейродегенеративных болезней накопления. Дебютирует в дошкольном (5–6 лет) или в школьном (7–10 лет) возрасте. Характеризуется манифестацией в виде генерализованных припадков (тонико-клонических судорог) или сложных парциальных (фокальных) приступов. По мере достижения пациентами пубертатного возраста частота приступов значительно уменьшается. В совершеннолетнем возрасте возможно достижение полной ремиссии по приступам [3, 5, 6, 9].
Катамениальная (менструальная) эпилепсия
При этой разновидности эпилепсии, не являющейся самостоятельной нозологической формой, возникновение приступов связано с фазами менструального цикла, подверженными влиянию многочисленных эндогенных и экзогенных факторов (предположительно, циклические изменения содержания в организме половых гормонов, нарушения водно-электролитного баланса, влияние полнолуния, колебания уровня антиэпилептических препаратов в крови). Для болезни характерна четкая зависимость от менструального цикла. По некоторым данным, среди девушек-подростков практически с равной частотой встречаются генерализованные формы болезни, ювенильная миоклонус-эпилепсия и ювенильная абсансная эпилепсия. Генерализованные судорожные пароксизмы обычно имеют тенденцию к учащению у всех пациентов с катамениальной эпилепсией [3, 5, 9].
Эпилепсии с точно не дифференцированным возрастным диапазоном дебюта
У части эпилепсий возрастные особенности дебюта считаются не определенными. Основные из них рассматриваются ниже.
Эпилепсия с миоклоническими абсансами
Болезнь чаще встречается в возрасте 5–10 лет и характеризуется клиническими проявлениями в виде абсансов, сочетающихся с интенсивными ритмичными двухсторонними клоническими или (реже) тоническими судорожными подергиваниями проксимальных мышц верхних и нижних конечностей, а также головы. Обычно сочетается с нарушениями психического развития и характеризуется фармакорезистентностью, что определяет малоблагоприятный прогноз болезни. Считается, что диагноз эпилепсии с миоклоническими абсансами устанавливается пациентам, соответствующим критериям детской абсансной эпилепсии, но при этом абсансные приступы должны сопровождаться миоклоническими подергиваниями. Поддается терапии вальпроатами, этосуксимидом или ламотриджином (сочетание вальпроатов с ламотриджином или этосуксимидом считается более эффективным); отмена лечения возможна не ранее чем через 2 года после достижения полной ремиссии (клинико-инструментальной) [3–5, 9].
Генерализованная эпилепсия с фебрильными судорогами плюс
Относится к генетически детерминированным эпилептическим синдромам и представляет собой несколько типов эпилепсии (GEFS+ тип 1, GEFS+ тип 2, GEFS+ тип 3, GEFS+ тип 5, ФС с афебрильными приступами и GEFS+ тип 7). Считается, что при GEFS+, впервые описанной в 1997 году, установление диагноза эпилепсии не является обязательным. Генерализованная эпилепсия с фебрильными судорогами плюс обычно отмечается у детей в возрасте 1–6 лет. Средний возраст детей к моменту дебюта GEFS+ cоставляет около 12 месяцев. Болезнь проявляется в виде фебрильных судорог на фоне лихорадки и в форме иных эпилептических пароксизмов. Помимо рецидивирующих фебрильных судорог (классических тонико-клонических), GEFS+ характеризуется наличием афебрильных приступов; клиническая картина болезни может включать абсансы, миоклонии, миоклонически-астатические и атонические припадки. В большинстве случаев фенотипы GEFS+ оказываются доброкачественными (по достижении подросткового возраста приступы чаще элиминируются). По достижении совершеннолетия у отдельных пациентов могут отмечаться редкие приступы (под влиянием стрессов и депривации сна). Обычно детям с GEFS+ не требуется антиэпилептической фармакотерапии, хотя некоторые авторы рекомендуют использовать бензодиазепины (в остром приступе или с превентивной целью). Назначение вальпроатов показано лишь в случаях, когда GEFS+ персистирует после 6-летнего возраста, а ламотриджин используется в случаях, резистентных к вальпроатам [3, 5, 6, 9].
Доброкачественная психомоторная эпилепсия детского возраста, или доброкачественная парциальная эпилепсия с аффективными симптомами
Локализационно-обусловленная очаговая форма эпилепсии. По этиологии бывает криптогенной, семейной или симптоматической. Встречается у детей различного возраста (7–17 лет). Доброкачественная психомоторная эпилепсия характеризуется рецидивирующими приступами, исходящими из очагов в височной доли, чаще всего из мезиальной области. Для болезни типичен широкий спектр психических феноменов, включая иллюзии, галлюцинации, дискогнитивные состояния и аффективные нарушения. Бóльшая часть сложных парциальных (фокальных) приступов исходит из височных долей. Для этой формы эпилепсии характерны моноформные приступы, начало в детском возрасте, возрастзависимое исчезновение всех клинических и ЭЭГ-проявлений [3–5, 9].
Атипичная доброкачественная парциальная эпилепсия, или синдром псевдо-Леннокса
Преимущественно отмечается у детей в возрасте 2–6 лет (74% наблюдений). Примерно у четверти детей к моменту дебюта болезни отмечаются признаки отставания в речевом развитии. У мальчиков дебютирует раньше, чем у девочек. Характеризуется генерализованными малыми приступами (атонически-астатические, миоклонические, атпичные абсансы). Отличительным признаком болезни является исключительно выраженная активация эпилептических приступов во время сна. Основным типом приступов являются малые генерализованные (67%), у 28% пациентов отмечаются простые парциальные приступы орофациальной области (или генерализованные тонико-клонические судороги, исходящие из орофациальной области). В дополнение к этому с различной частотой у детей встречаются следующие типы припадков (в порядке убывания): генерализованные тонико-клонические (44%), парциальные моторные (44%), односторонние (21%), версивные (12%), фокальные атонические (9%), сложные парциальные (2%). У незначительной части пациентов отмечается феномен эпилептического негативного миоклонуса. ЭЭГ-картина напоминает таковую при роландической эпилепсии (очаговые острые медленные волны и пики), но характеризуется генерализацией во время сна. В отношении приступов прогноз болезни благоприятен (все пациенты к 15-летнему возрасту «свободны от приступов»), но нередко у детей отмечается интеллектуальный дефицит различной степени выраженности (около 56% наблюдений) [3, 5].
Синдром Айкарди
Разновидность эпилептического синдрома, ассоциированного с мальформациями ЦНС (нарушения корковой организации, шизэнцефалия, полимикрогирия. Синдром, описанный J. Aicardi и соавт. (1965), включает агенезию мозолистого тела с хориоретинальными нарушениями и характеризуется инфантильными флексорными спазмами. Поражает почти исключительно девочек, хотя известны 2 случая регистрации болезни у мальчиков с аномальным генотипом (оба ребенка имели по две Х-хромосомы). Помимо эпилептических проявлений, для синдрома Айкарди типичны следующие патологические изменения: хориоретинальные лакунарные дефекты, полная или частичная агенезия мозолистого тела, пороки развития грудного отдела позвоночника, микрофтальмия, колобома зрительного нерва и др. Клинически синдром Айкарди характеризуется инфантильными спазмами (нередко с ранним дебютом) и парциальными (фокальными) эпилептическими приступами (в первые дни-недели жизни), а также выраженным отставанием в интеллектуальном развитии. Эпилептические приступы при синдроме Айкарди практически всегда оказываются фармакорезистентными. Прогноз заболевания неблагоприятен [3, 5–7, 9].
Электрический эпилептический статус медленно-волнового сна (ESES)
Эта разновидность эпилепсии известна также под другим названием (эпилепсия с постоянными пик-волновыми разрядами во время медленного сна, CSWS). Считается идиопатической эпилепсией и дебютирует у детей примерно с 2-летнего возраста. Клинически характеризуется фокальными, генерализованными тонико-клоническими и/или миоклоническими приступами, которые возникают в состоянии бодрствования или сна (наблюдаются не всегда). Впоследствии болезнь приводит к нарушениям речевого развития, расстройствам поведения, когнитивным дисфункциям различной выраженности. Диагноз устанавливается на основании данных ЭЭГ во время сна (специфический паттерн в виде непрерывной генерализованной пик-волновой активности); при этом эпилептиформная активность должна занимать 85–100% от общей продолжительности фазы медленного сна. В момент пробуждения ЭЭГ позволяет зарегистрировать наличие острых волн. Прогноз в плане исчезновения эпилептических приступов и ЭЭГ-изменений относительно благоприятен (к пубертатному периоду), но у детей сохраняются нарушения когнитивных функций [3–6].
Лобная ночная аутосомно-доминантная эпилепсия
Относится к изолированным эпилептическим синдромам. Дебютирует в возрасте до 20 лет (чаще примерно в 11-летнем возрасте). Припадки возникают при засыпании и/или просыпании (в виде коротких — до 1 мин, эпизодов гиперкинезов, с потерей сознания или без такового); эпилептическим приступам предшествует аура (ощущение страха, дрожь или соматосенсорные феномены). У 50–60% пациентов наблюдаются вторично-генерализованные судорожные припадки; примерно в четверти случаев приступы происходят в период бодрствования.
Иктальное ЭЭГ-исследование регистрирует острые и медленные волны или ритмичную низковольтажную быструю активность в лобных отведениях. В межприступном периоде данные ЭЭГ могут быть нормальными или периодически обнаруживать пики в лобных отведениях [3, 5, 9].
Заключение
Среди эпилепсий, встречающихся у детей любого возраста (0–18 лет), необходимо перечислить кожевниковскую эпилепсию (хроническая прогрессирующая парциальная эпилепсия, или epilepsia partialis continua), клинические проявления которой хорошо знакомы детским неврологам, а также локализационно-обусловленные формы эпилепсии (лобная, височная, теменная, затылочная) [3]. Последние относятся к симптоматическим и вероятно симптоматическим фокальным эпилепсиям, при которых широкий спектр симптоматики определяется локализацией эпилептогенного очага.
Литература
- Броун Т. Р., Холмс Г. Л. Эпилепсия. Клиническое руководство. Пер. с англ. М.: Изд-во БИНОМ. 2006. 288 с.
- Мухин К. Ю., Петрухин А. С. Идиопатические формы эпилепсии: систематика, диагностика, терапия. М.: Арт-Бизнес-Центр, 2000. 319 с.
- Эпилепсия в нейропедиатрии (коллективная монография) / Под ред. Студеникина В. М. М.: Династия, 2011, 440 с.
- Child neurology (Menkes J. H., Sarnat H. B., Maria B. L., eds.). 7 th ed. Lippincott Williams&Wilkins. Philadelphia-Baltimore. 2006. 1286 p.
- Epileptic syndromes in infancy, childhood and adolescence (Roger J., Bureau M., Dravet Ch., Genton P. et al, eds.). 4 th ed. (with video). Montrouge (France). John Libbey Eurotext. 2005. 604 p.
- Encyclopedia of basic epilepsy research/Three-volume set (Schwartzkroin P., ed.). 1–3. Philadelphia. Elsevier/Academic Press. 2009. 2496 p.
- Aicardi J. Diseases of the nervous system in children. 3 rd ed. London. Mac Keith Press / Distributed by Wiley-Blackwell. 966 p.
- Chapman K., Rho J. M. Pediatric epilepsy case studies. From infancy and childhood through infancy. CRC Press / Taylor&Francis Group. Boca Raton–London. 2009. 294 p.
- Epilepsy: A comprehensive textbook (Engel J., Pedley T. A., eds.). 2 nd ed. Vol. 1–3. Lippincott Williams&Wilkins/A Wolters Kluwer Business. 2986 p.
Источник: Журнал «Лечащий врач» №1 2015
Клиническая диагностика эпилепсии основывается на сборе анамнеза, осмотре пациента (неврологическом и общесоматическом), проведении рутинных параклинических исследований (лабораторных и инструментальных) [1].
Диагностика эпилепсии у детей
Предлагается рассматривать следующие этапы установления диагноза при эпилепсии у детей: 1) описание пароксизмального события (возможно по данным анамнеза); 2) классификация приступа (анамнез, визуальное наблюдение, электроэнцефалография (ЭЭГ)); 3) диагностика формы эпилепсии (клинические данные + ЭЭГ + методы нейровизуализации); 4) установление точного диагноза (магнитно-резонансная томография (МРТ), кариотип); 5) диагностика сопутствующих заболеваний и установление степени инвалидизации [1–3].
Сбор анамнеза осуществляется как у родителей больного, так и у самого пациента (при наличии у ребенка способности к адекватному вербальному общению). Особое внимание при сборе анамнеза придается следующим моментам: возраст больного к моменту первого приступа (ранняя манифестация болезни обычно характеризуется ее более тяжелым течением); наследственность (наличие эпилепсии у родителей, прародителей, сибсов, ближайших родственников; наличие у них же иных пароксизмальных расстройств церебральных функций); наличие в анамнезе неонатальных приступов (судорог) как факторов риска по развитию эпилепсии; течение беременности данным ребенком (осложнения гестационного и родового периодов); наличие в анамнезе фебрильных судорог (фактор риска); перинатальная патология (поражение центральной нервной системы (ЦНС) гипоксического, ишемического, травматического и инфекционного генеза, токсические воздействия); гипoксические, ишемические, токсические, травматические или инфекционные поражения ЦНС, отмечавшиеся по завершении неонатального периода; наличие cопутствующих соматических или психоневролагических заболеваний; прием лекарственных средств, обладающих способностью индуцировать судороги или эпилептические приступы (в прошлом или в настоящее время); для подростков — вредные привычки (табакокурение, употребление алкоголя), токсикомания, прием психоактивных веществ; при приеме антиэпилептических препаратов — их переносимость; у девочек, достигших периода пубертата, — наличие/отсутствие связи приступов с менструальным циклом. Для пациентов, достигших определенного возраста (пубертат), необходима характеристика течения заболевания с учетом его возрастной эволюции (периоды гормональной перестройки); такой подход позволяет выявить влияние гормонального фона на течение эпилепсии [1, 4].
При этом следует собрать детальную информацию относительно самих приступов, в частности, значение придается следующим основным факторам: частота приступов (за сутки, неделю, месяц, год); продолжительность приступов (в минутах); наличие/отсутствие у пациента постиктального паралича/пареза Тодда и его продолжительность; время типичного возникновения приступов (утро, день, вечер, ночь); связь приступов с засыпанием и/или сном; наличие/отсутствие ауры; факторы, предрасполагающие к эпилептическим приступам (недосыпание, психоэмоциональный стресс, избыточные физические и интеллектуальные нагрузки, употребление конкретных фармакопрепаратов или интоксикантов и т. д.); психоневрологическая симптоматика во время приступа; индивидуальные особенности приступов, типичные для конкретного пациента [1, 5].
Оценка неврологического статуса у детей с установленной или предполагаемой эпилепсией проводится в полном объеме. Неврологический осмотр нацелен на выявление возможных очаговых (фокальных) нарушений функций головного мозга, а также выраженности психоневрологического дефицита [1].
Общий анализ крови и мочи обязателен при обследовании детей с эпилепсией (до начала лечения и после его инициации). Cодержание гемоглобина, количество лейкоцитов и тромбоцитов в крови определяют в целях исключения фолиеводефицитной анемии, а также ассоциированных с этой патологией вторичных изменений со стороны костного мозга, могущих проявляться в виде снижения уровня лейкоцитов (лейкопения) и тромбоцитов (тромбоцитопения). Определение относительной плотности мочи производится для оценки почечных функций и исключения сопутствующей ренальной патологии (почечная недостаточность и т. д.), требующей последующей коррекции дозировки используемых антиэпилептических препаратов [1, 6].
Электроэнцефалографическое исследование — основной электрофизиологический метод, используемый в диагностике эпилепсии. Целью ЭЭГ-исследования при эпилепсии является выявление типичных (специфических) электрофизиологических признаков, соответствующих той или иной форме болезни [1, 7]. При некоторых формах эпилепсии у детей, когда данные обычного ЭЭГ-исследования оказываются недостаточно информативными, показано проведение видео-ЭЭГ-мониторинга в период бодрствования и сна (продолжительная регистрация ЭЭГ с одновременной видеозаписью — в течение 1,5–24 часов). Вероятность регистрации у ребенка патологической (эпилептиформной) электрической активности при засыпании или во время сна существенно возрастает [2, 7]. Видео-ЭЭГ-мониторинг позволяет получить не только паттерны биоэлектрической активности мозга, но и обеспечивает значительный объем дополнительной информации (мимика, состояние тонкой и грубой моторики во время приступа и вне его). Синхронизация ЭЭГ-исследования и видеозаписи дает возможность объективизировать имеющиеся у пациента пароксизмальные нарушения различного генеза (эпилептического и неэпилептического). Для видео-ЭЭГ-мониторинга требуется специальная аппаратура. Отсутствие у пациента эпилептической активности при ЭЭГ-исследовании не является основанием для отрицания наличия у него эпилепсии [8].
Методы нейровизуализации предусматривают не только компьютерную томографию (КТ) и МРТ головного мозга, но и проведение эхоэнцефалографическое исследование (Эхо-ЭГ) головного мозга (двухмерное) — в периоде новорожденности и на протяжении первого года жизни (через большой родничок, в двух основных плоскостях — коронарной/фронтальной и сагиттальной/парасагиттальной). Одномерная Эхо-ЭГ применяется у детей любого возраста для оценки состояния мозга при подозрении на наличие объемного образования, гидроцефалии, кровоизлияния и т. д. (условность эхо-сигналов и одномерность эхограммы резко ограничивают диагностическую ценность последнего исследования) [1].
Диффузионное МРТ-исследование — один из новых видов МРТ-определения «каналов» белого вещества в глубоких слоях мозга (их обнаружение позволяет предотвратить развитие неврологического дефицита при нейрохирургическом вмешательстве). Функциональное МРТ-исследование (фМРТ) — прогрессивный метод нейровизуализации, определяющий функционально значимые участки мозга (речевые центры и моторные зоны), являющиеся индивидуальными для каждого пациента. Даже при идиопатических генерализованных эпилепсиях, обычно не сопровождающихся патологическими изменениями при МРТ-исследовании, фМРТ позволяет в ряде случаев обнаружить стуктурные и/или функциональные аномалии. При детской абсансной эпилепсии одновременная регистрация данных фМРТ и ЭЭГ обнаруживает у пациентов электрографические вспышки пик-волновой активности, ассоциированные с интенсивной билатеральной активацией таламуса, зависящей от уровня оксигенации крови. При доброкачественной эпилепсии с центротемпоральными пиками одновременное применение фМРТ/ЭЭГ выявляет очаговую активацию в роландической области (с высокой степенью специфической локализации — при помощи этих исследований) [1, 4].
Содержание в крови аспартат-трансаминазы и аланин-трансаминазы определяют для выявления гепатотоксичного эффекта, свойственного некоторым антиэпилептическим препаратам; активность щелочной фосфатазы и γ-глутаминтрансферазы в крови считается в отношении гепатотоксичности более чувствительными показателями. Исследование уровня креатинина в крови и изучение клубочковой фильтрации производится при подозрении на наличие у пациента почечной недостаточности. Другие биохимические показатели, исследуемые в крови у детей и подростков с эпилепсией в различных клинических ситуациях, довольно многочисленны: глюкоза, мочевина, креатинфосфокиназа, содержание кальция (общего и ионизированного), фосфаты неорганические, калий, натрий, магний, хлор, железо, лактатдегидрогеназа, aммиак, общее содержание белка и протеинограмма, альбумин, амилаза, молочная кислота, азот мочевины и др. [1, 6].
Оценка уровня стероидных половых гормонов (пролактин, эстрогены) у детей с эпилепсией производится редко (преимущественно у девочек-подростков, достигших пубертата). В ряде случаев указанное исследование позволяет оптимизировать проводимое антиэпилептическое лечение (определенные гормоны могут обладать проконвульсивным или антиконвульсивным действием) [1, 5].
При необходимости выполняются иммунологические исследования (изучение показателей клеточного и гуморального иммунитета), нейрохимические исследования (тест пароксизмальной активности, тест ишемии и др.), патопсихологические исследования (батарея психологических тестов: MMSE, MSQ, OMC test, 7MSI, CD test, Mattis DRS, DAS, WPPSI-III и многие другие) [9].
Так называемый Wada-тест (селективный интракаротидный амобарбиталовый тест) применяется для выявления у пациента доминантного полушария (латерализации функций речи и памяти с целью избежания когнитивного дефицита, например, при нейрохирургическом вмешательстве на структурах височной доли) [10].
Для объективизации когнитивных функций у детей с эпилепсией используются тестовые компьютерные системы (Психомат и др.) [1, 11].
В детской эпилептологии все активнее применяются методы молекулярной, биохимической и клинической генетики (газожидкостная и высокоэффективная жидкостная хроматография, хромато-масс-спектрометрия, цитогенетический и молекулярно-генетический анализ и т. д.). Среди нейрогенетических методов исследований следует отметить составление родословных (определение типа наследования, риска рождения больного ребенка, пенетрантность и экспрессивность того или иного гена); цитогенетические методы (определение числа и строения хромосом); картирование патологических генов и митохондриальных генов и т. д. [1, 5].
Фармакомониторинг содержания антиэпилептических препаратов в крови нацелен на поддержание оптимальной концентрации в организме пациентов этих лекарственных средств, обладающих сравнительно узким спектром фармакологической направленности. Оптимальному терапевтическому эффекту обычно соответствует определенная средняя концентрация (или диапазон концентраций) антиэпилептических препараов в крови («терапевтический коридор»). Чаще всего при проведении фармакомониторинга антиэпилептических препаратов используется плазма крови, хотя возможно определение их концентрации и в других физиологических жидкостях (спинномозговая жидкость, моча, слюна и др.). Исследование фармакокинетики антиэпилептических препаратов позволяет максимально индивидуализировать и оптимизировать проводимую терапию у детей (дозировка, кратность приема, профилактика нежелательных лекарственных реакций (НЛР)). Для фармакомониторинга применяются специальные автоматические анализаторы [1, 5, 12].
Среди других лабораторных методов исследований, применяемых при обследовании пациентов с эпилепсией, следует перечислить следующие: исследование спинномозговой жидкости (полученной в результате спинальной пункции), кальций-креатининовый коэффициент в моче, дополнительные иммунологические показатели (циркулирующие иммунные комплексы, фагоцитарная активность нейтрофилов, комплемент и его фрагменты, функциональные показатели компонентов комплемента в плазме крови — общая гемолизирующая активность и уровень общего распада, компоненты классического и альтернативного пути активации комплемента в сыворотке крови — C1q, C1r, C1s, C2, C3, C4, C5, C6, C7 и C4-связывающий белок, фактор В, нефелометрия, пропердин, β1H-глобулин, ингибитор С1, инактиватор С3b, белок S; реакция бластной трансформации лимфоцитов, фитогемагглютинин, конканавалин-А, митоген лаконоса, спонтанный и индуцированный индекс супрессии, иммунорегуляторный индекс, цитокиновый статус — цитокины и фактор некроза опухоли (ФНО), НСТ-тест, содержание антител в спинномозговой жидкости и др.); определение интерферонового статуса; исследование нейрон-специфической енолазы; количественный анализ содержания в плазме крови оксида азота (NO) и его стойких метаболитов (ионы NO2- и NO3-); анализ содержания в моче и/или крови амино- и органических кислот; определение содержания кетоновых тел в моче (при использовании кетогенных диет) и др. [1, 6, 13].
Инструментальные методы диагностики, требующие применения в тех или иных клинических ситуациях, включают исследование зрительных вызванных потенциалов (на вспышку и «шахматный» паттерн) — у пациентов с парциальной (фокальной) эпилепсией; магнитно-резонансную спектроскопию; околоинфракрасную спектроскопию; спонтанную протонно-эмиссионную томографию и протонно-эмиссионную томографию; однофотонно-эмиссионную компьютерную томографию; транскраниальную допплерографию; ультразвуковую допплерографию; транскраниальную магнитную стимуляцию; электрокардиографию; холтеровский ЭКГ-мониторинг; Эхо-кардиографию; сцинтиграфию (перфузионную, динамическую, статическую) головного мозга; церебральную ангиографию; иридодиагностику (иридоскопию и иридографию); аурикулодиагностику; сомнологические (полисомнографические) исследования и др. [5, 12].
Лечение эпилепсии у детей — сложная и комплексная задача. Основой терапии эпилепсии является назначение антиэпилептических препаратов, а целью лечения — предотвращение развития судорожных и бессудорожных припадков и ассоциированных с ними когнитивных нарушений [1].
Особенностью эпилепсии можно считать то обстоятельство, что при лечении этого заболевания необходим длительный (многолетний) прием препаратов, препятствующих развитию припадков (не менее двух лет после полного прекращения эпилептических приступов).
Антиэпилептические препараты, используемые в неврологии детского возраста, в основном соответствуют средствам, применяемым при лечении эпилепсии у взрослых, хотя имеется ряд ограничений и противопоказаний к активному назначению отдельных препаратов.
Принципы лечения эпилепсии у детей
Наличие у ребенка более одного клинически и/или инструментально подтвержденного эпилептического приступа (нефебрильного) является прямым показанием для инициации соответствующего лечения. Сложнее дело обстоит с ситуациями, когда у ребенка отмечался первый приступ, что по определению еще не может расцениваться как эпилепсия, или приступов вообще не было, но имеются специфические изменения на электроэнцефалограмме [1].
Существует определенная дискутабельность в подходах к лечению пациентов, у которых отмечался всего один (первый) приступ. Основным моментом в принятии решения о начале антиэпилептической терапии является объективная оценка риска повторных приступов. Для такой оценки необходимы неврологический осмотр пациента, тщательный сбор анамнеза (наличие указаний на эпилепсию у ближайших родственников, перенесение черепно-мозговой травмы или нейроинфекций, описание имевшегося у пациента приступа и постприступного периода и т. д.) в совокупности с инструментально-лабораторными методами исследований (данные ЭЭГ, МРТ, биохимического анализа крови и др.). Вполне естественно, что в подавляющем большинстве случаев дебют болезни в виде эпилептического статуса является показанием к началу антиэпилептической терапии, как и непровоцированное развитие первого приступа [1].
В систематическом обзоре J. J. Shih и J. G. Ochoa (2009) указывается, что «назначение антиэпилептических препаратов после первого приступа снижает риск повторного рецидива болезни в ближайшем периоде, но не оказывает благоприятного влияния на отдаленный прогноз развития эпилепсии» [14]. W. F. Arts и A. T. Geerts (2009) считают возможным воздержаться от немедленного проведения детям антиэпилептического лечения после первого приступа, при непровоцированном эпилептическом статусе, а также при неоднократных, но нечастых эпилептических приступах [15].
Современная концепция эпилептологии, поддерживаемая положениями доказательной медицины, подразумевает, что принятие решения об инициации антиэпилептической терапии должно быть строго индивидуализированным и основанным на адекватной оценке риска возникновения у конкретного пациента повторных приступов (с последующей инвалидизацией); при этом также обязательно следует учитывать риск физических, когнитивных и психологических нежелательных лекарстенных реакций (НЛР), ассоциированных с применением противоэпилептических средств (баланс терапевтического и антитерапевтического эффектов) [1, 5, 12].
Что касается возможности инициации противоэпилептического лечения на основании данных одних лишь нейроинструментальных методов исследований (ЭЭГ, МРТ) — при отсутствии приступов у пациента, то подобный подход следует признать неоправданным и чрезвычайно рискованным в связи с высокой вероятностью формирования у ребенка соматоневрологической патологии, ассоциированной с нежелательными лекарственными реакциями антиэпилептических препаратов [1].
Для подбора адекватного противоэпилептического средства (или их сочетания) требуются сведения не только о клинических особенностях приступа, которые могут выражаться в сложной (клинической) симптоматологии, но и о динамике течения заболевания.
Имеет значение механизм действия антиэпилептического препарата и место его приложения, а также взаимодействие с другими лекарственными средствами. К сожалению, механизмы действия подавляющего большинства антиэпилептических препаратов, несмотря на многочисленные исследования, не могут считаться окончательно изученными [5].
Среди антиэпилептических препаратов I поколения только вальпроаты обладают доказанной эффективностью при всех типах эпилептических приступов, не вызывая отрицательного эффекта в виде аггравации приступов у пациентов. Из новых антиэпилептических препаратов (II поколения) мало какое средство может претендовать на роль заменителя вальпроатов.
При проведении фармакологического лечения эпилепсии у пациентов детского возраста рекомендуется назначение адекватной для данных типов припадков и эпилептических синдромов терапии одним из препаратов первого ряда. Лечение начинают с небольшой дозы и постепенно увеличивают ее до прекращения припадков или появления признаков передозировки. Примерно у 70% пациентов правильно подобранная монотерапия обеспечивает адекватный контроль припадков [1, 5].
Фармакорезистентность — серьезная проблема в эпилептологии (около 30–40% случаев эпилепсии в детском возрасте рефрактерны к лечению, а некоторые виды эпилепсии у детей фармакорезистентны по определению). К случаям рефрактерной эпилепсии в детском возрасте следует относить те ситуации, когда болезнь не поддается медикаментозному контролю, несмотря на адекватную попытку применения трех антиэпилептических препаратов первой линии выбора, а также сопровождается нарушениями нормального развития и/или препятствует нормальной жизни ребенка [1, 16].
По предложению M. J. Brodie и S. C. Schachter (2005) выделяют ряд комбинаций, которые считаются эффективными и рекомендуются при фармакорезистентных эпилепсиях: вальпроаты + этосуксимид, карбамазепин + вальпроаты, вальпроаты + ламотриджин, вигабатрин* + ламотриджин, вигабатрин + тиагабин*, топирамат + ламотриджин [17].
Препаратами выбора при фокальных припадках (без вторичной генерализации или вторично-генерализованных) считаются вальпроаты или карбамазепин. При отдельных формах эпилепсии к препаратам первого выбора относятся новые антиэпилептические средства (топирамат, ламотриджин).
При генерализованных приступах (первично-генерализованных тонико-клонических, абсансах, миоклонических) препаратами выбора являются вальпроаты и топирамат; карбамазепин и фенитоин противопоказаны при абсансах и миоклонических припадках. При простых абсансах препаратами выбора являются вальпроаты или этосуксимид [1, 16].
Атипичные абсансы, атонические и тонические припадки зачастую оказываются резистентными к лечению. В индивидуальных случаях могут быть эффективны фенитоин, вальпроаты, ламотриджин, клоназепам, этосуксимид, фенобарбитал, ацетазоламид и глюкокортикоиды или их сочетание [1, 16].
При миоклонических припадках препаратом выбора является вальпроат натрия, применяют также клоназепам, ламотриджин. При недостаточной эффективности или плохой переносимости традиционных антиэпилептических препаратов применяют новые антиконвульсанты (например, ламотриджин или топирамат) [1, 16, 18].
При недифференцированных припадках следует применять препараты широкого спектра действия (вальпроаты, топирамат и др.). Только в случаях неэффективности правильно подобранной монотерапии (после не менее чем двух последовательных попыток применения препаратов в режиме монотерапии) возможно использование политерапии (речь идет о препаратах первого выбора, считающихся адекватными для конкретного типа эпилептических припадков). Длительное лечение двумя препаратами осуществляют исключительно при невозможности проведения адекватной монотерапии. Возможна постепенная замена первого дополнительного препарата (в случае его неэффективности) другим дополнительным препаратом. Лечение тремя препаратами целесообразно только при неэффективности терапии двумя адекватными препаратами [1, 15].
Практически все НЛР, возникающие у детей и подростков на фоне лечения антиэпилептическими препаратами, можно классифицировать по трем основным категориями: 1) дозозависимые эффекты; 2) реакции гиперчувствительности и идиосинкратические (крайне редкие). Кроме того, выделяют ранние (возникающие в течение первых недель терапии) и поздние (проявляющиеся через несколько месяцев-лет) НЛР. В целях профилактики и коррекции основных НЛР большинства антиэпилептических препаратов рекомендуется использование режима монотерапии; регулярный фармакомониторинг содержания препаратов в крови; применение пролонгированных лекарственных форм; временное снижение дозы используемого антиэпилептического препарата; альтернативные немедикаментозные методы антиэпилептического лечения; симптоматическая терапия проявлений со стороны различных систем и органов организма, возникающих вследствие НЛР антиэпилептических препаратов [1, 18].
Отмена антиэпилептической терапии всегда должна быть постепенной, с обязательным учетом формы эпилепсии и ее прогноза, возможности возобновления припадков, индивидуальных и возрастных особенностей ребенка. Отмену противоэпилептической терапии проводят, как правило, не менее чем через 2–3 года после полного прекращения припадков (рекомендуют также и срок до 5 лет), под контролем данных ЭЭГ-исследования [1, 5, 18].
Частота приема антиэпилептических препаратов определяется их временем полувыведения. Во всех случаях следует стремиться к минимально допустимой кратности приема конкретных препаратов (не более 2 раз в день). По современным представлениям, весьма целесообразным выглядит использование пролонгированных форм антиэпилептических препаратов. Количество НЛР при назначении детям пролонгированных форм препаратов существенно уменьшается по сравнению с использованием их обычных (традиционных) форм, отмечается их лучшая переносимость и более высокая клиническая эффективность — преимущественно за счет достижения стабильной концентрации препаратов в плазме крови [1, 5].
Фармакомониторинг необходим при использовании фенитоина, карбамазепина, препаратов вальпроевой кислоты, фенобарбитала, этосуксимида, примидона. Для большинства вышеупомянутых антиэпилептических препаратов выявлены статистически значимые корреляции между терапевтической эффективностью этих лекарственных средств и их концентрацией в крови больных [5, 12].
Необходимо учитывать особенности фармакокинетики и фармакодинамики различных антиэпилептических препаратов у пациентов в возрасте 0–18 лет. Трудности фармакотерапии эпилептических синдромов у детей первых лет жизни сопряжены с этиологией эпилепсии, наличием сопутствующей патологии, возможностью взаимодействия антиэпилептических препаратов с другими средствами, принимаемыми пациентами для лечения соматических состояний, а также с возрастными особенностями абсорбции и метаболизма лекарственных средств. Особенностью использования антиэпилептических препаратов в детской неврологии является необходимость рутинного проведения профилактических и корригирующих мероприятий, позволяющих избежать НЛР при применении антиконвульсантов в составе моно- и политерапии [1, 5].
Когнитивные дисфункции связаны как с течением эпилептического процесса, так и с реализацией НЛР антиэпилептических препаратов. Поскольку нарушения когнитивных функций отмечаются у 25–30% пациентов с эпилепсией, необходима их коррекция [1, 11]. Современная концепция комплексного подхода к терапии эпилепсии, поддерживаемая A. P. Aldenkamp (2001), гласит, что лечение эпилепсии не может считаться адекватным, если нацелено исключительно на элиминацию или уменьшение числа эпилептических приступов и игнорирует когнитивные аспекты болезни [19].
В этой связи в коррекции когнитивных нарушений у пациентов с эпилепсией используют ноотропы, нейрометаболиты, сосудистые средства, аминокислотные препараты; по показаниям используются комплекс психотерапевтических мероприятий, метод биологической обратной связи и др. [1, 11].
- A. Ketter и соавт. (1999), анализируя позитивные и негативные эффекты антиэпилептических препаратов, высказали гипотезу, которой в настоящее время придерживаются многие исследователи [20]. В соответствии с гипотезой Кеттера, антиэпилептические препараты обладают различными механизмами действия, поэтому у них отмечаются разнообразные эффекты (противосудорожные, психотропные и побочные). На основании их психотропных профилей были выделены две основные категории препаратов: 1) с так называемым «седативным» спектром (с которым ассоциируются утомляемость, угнетение когнитивных функций, а также анксиолитическое и антиманиакальное действие); 2) cо «стимулирующим» спектром действия (стимуляция, улучшение настроения, антидепрессивный эффект) [20].
Эффекты препаратов седативного спектра могут быть связаны с потенцированием ГАМК-тормозящих нейротрансмиттерных систем и вызываются барбитуратами, бензодиазепинами, вальпроатами, габапентином, тиагабином и вигабатрином. Действие антиэпилептических препаратов стимулирующего спектра ассоциировано с подавлением глутаматергических возбуждающих нейротрансмиттерных систем; к ним относятся фелбамат* и ламотриджин. Топирамат, обладающий ГАМКергическим и антиглутаматергическим механизмами действия, может иметь «смешанные» профили [1, 16].
Гипотеза Кеттера предполагает, что более благоприятный психиатрический исход у пациентов с эпилепсией может быть достигнут при назначении «седативных» ГАМКергических препаратов больным с нарушениями возбуждающего спектра (бессонница, ажитация, выраженное беспокойство, потеря веса и др.) и, наоборот, стимулирующих антиглутаматергических препаратов — пациентам с выраженной заторможенностью или явлениями астенизации (с гиперсомнией, апатией, депрессией, замедлением мышления и др.) [20].
В свою очередь, нарушения поведения у пациентов с эпилепсией также могут индуцироваться как течением самой болезни, так и быть следствием ятрогенных воздействий (антитерапевтический эффект противоэпилептических средств и др.) [11]. При лечении нарушений поведения (тревожность, депрессия, невротические реакции и т. д.) детским неврологам важно четко определить момент, когда для выработки и осуществления адекватных терапевтических мероприятий ребенку требуется помощь со стороны психиатра (при возникновении синдромов изменения сознания, синдромов дереализации, психической расторможенности, суицидальных мыслях или поведении и т. д) [1, 5].
Лечение эпилептических приступов и коррекция когнитивных нарушений не исчерпывают терапевтических возможностей при эпилепсии. При этой группе болезней предусмотрена (по показаниям) симптоматическая терапия нарушений мышечного тонуса, тазовых расстройств, координаторных нарушений, повышенной утомляемости и т. д. Значительная часть патологических состояний, сопутствующих эпилепсии, является прямым следствием антитерапевтического эффекта используемых противоэпилептических средств (остеопенические состояния, поражение печени, почек и желудочно-кишечного тракта, нарушения со стороны сердечно-сосудистой системы, эндокринных органов и т. д.). Описываемые состояния требуют проведения соответствующей симптоматической терапии, возможности которой не следует игнорировать [1, 5, 12].
Метаболическая терапия используется при эпилепсиях, являющихся следствием врожденных нарушений обмена. N. I. Wolf и соавт. (2005) указывают на эффективность метаболической терапии при эпилепсии вследствие недостаточности GLUT1 (транспортера глюкозы 1) (кетогенные диеты); эпилепсии вследствие дефектов биосинтеза серина (дотация серина); кофактор-зависимой эпилепсии (пиридоксин, пиридоксальфосфат, фолиевая кислота, биотин); эпилепсии вследствие дефицита GAMT (гуанидиноацетат метилтрансферазы) (дотация креатина, диета с ограничением аргинина и обогащением орнитином); эпилепсии при фенилкетонурии (ФКУ) (низкофенилаланиновая диета) [21]. При атипичных формах ФКУ используется заместительная терапия (L-допа, 5ОН-триптофан, фолиевая кислота). Перечисленные разновидности метаболической терапии могут использоваться по строгим медицинским показаниям при условии точного установления диагноза эпилепсии, обусловленной теми или иными врожденными нарушениями метаболизма [21, 22].
Литература
- Эпилепсия в нейропедиатрии (коллективная монография) / Под ред. Студеникина В. М. М.: Династия. 2011, 440 с.
- Мухин К. Ю., Петрухин А. С., Глухова Л. Ю. Эпилепсия. Атлас электроклинической диагностики. М.: Альварес Паблишинг. 2004. 440 с.
- Arzimanoglou A., Guerrini R., Aicardi J. Aicardi’s epilepsy in children. 3 rd ed. Philadelphia-Tokyo. Wolters Kluwer. 2004. 516 p.
- Chapman K., Rho J. M. Pediatric epilepsy case studies. From infancy and childhood through infancy. CRC Press/Taylor&Francis Group. Boca Raton–London. 2009. 294 p.
- Engel J., Pedley T. A. eds. Epilepsy: A comprehensive textbook 2 nd ed. 1–3. Lippincott Williams&Wilkins/A Wolters Kluwer Business. 2008. 2986 p.
- Gilbert-Barness E., Barness L. A. (eds). Clinical use of pediatric diagnostic tests. Philadelphia-Baltimore. Lippincott Williams & Wilkins. 2003. 924 p.
- Зенков Л. Р. Клиническая эпилептология (с элементами нейрофизиологии). М.: Медицинское информационное агентство. 2002. 416 с.
- Айвазян С. О., Ширяев Ю. С. Видео-ЭЭГ-мониторинг в диагностике эпилепсии у детей // Ж. неврол. психиатрии им. С. С. Корсакова. 2010. Т. 110. № 6. С. 70–76.
- Strauss E., Sherman E. M. S., Spreen O. (eds). A Compendium of neuropsychological tests (Administration, norms, and commentary) 3 rd ed. 2006. Oxford University Press. Oxford-New York. 1216 p.
- Wada J., Rasmussen T. Intracarotid injection of sodium amytal for the lateralization of cerebral speech dominance. 1960 // J. Neurosurg. 2007. Vol. 106. P. 1117–1133.
- Дзюба С. В. Состояние когнитивных функций при эпилепсии у детей. Автореф. дис. … к.м.н. М., 1998. 26 с.
- Encyclopedia of basic epilepsy research/Three-volume set (Schwartzkroin P., ed.). 1–3. Philadelphia. Elsevier/Academic Press. 2009. 2496 p.
- Студеникин В. М., Балканская С. В., Звонкова Н. Г., Высоцкая Л. М. и др. Маркеры иммунных процессов при эпилепсии у детей // Вопр. совр. педиатрии. 2006. Т. 5. № 1. С. 550–551.
- Shih J. J., Ochoa J. G. A systematic review of antiepileptic drug initiation and withdrawal // Neurologist. Vol. 15. P. 122–131.
- Arts W. F., Geerts A. T. When to start drug treatment for childhood epilepsy: the clinical-epidemiological evidence // Eur. J. Paediatr. 2009. Vol. 13. P. 93–101.
- Shorvon S., Perucca E., Engel J. Jr. (eds). The treatment of epilepsy. 3 rd ed. Chichester (UK). Wiley-Blackwell/A John Wiley & Sons, Ltd. 2009. 1076 p.
- Brodie M. J., Schachter S. C. Epilepsy: fast facts. 3 rd ed. Oxford. Health Press Limited. 2005, 127 p.
- Roger J., Bureau M., Dravet Ch., Genton P. et al., eds. Epileptic syndromes in infancy, childhood and adolescence 4 th ed. (with video). Montrouge (France). John Libbey Eurotext. 2005. 604 p.
- Aldenkamp A. P. Effects of antiepileptic drugs on cognition // Epilepsia. Vol. 42. Suppl. 1. S. 46–49.
- Ketter T. A., Post R. M., Theodore W. H. Positive and negative psychiatric effects of antiepileptic drugs in patients with seizure disorders // Neurology. Vol. 53. S. 53–67.
- Wolf N. I., Bast T., Surtees R. Epilepsy in inborn errors of metabolism // Epileptic Disord. Vol. 7. P. 67–81.
- Walsh L. E., McCandless D. Inherited epilepsies // Sem. Neurol. 2001. Vol. 8. P. 165–176.

Комментировать