Общая характеристика эпилепсии: классификация заболевания. Неклассифицируемые эпилептические припадки. Этиология и патогенез эпилепсии.
В. М. Студеникин , доктор медицинских наук, профессор, член-корреспондент РАЕ, ФГБУ «НЦЗД» РАМН, Москва
Эпилепсия — общее название группы хронических пароксизмальных болезней головного мозга, проявляющихся повторными судорожными или другими (бессудорожными) стереотипными припадками, сопровождающихся разнообразными (патологическими) изменениями личности и снижением когнитивных функций.
Детская неврология, в которой постоянно совершенствуются представления о метаболических, иммунологических и молекулярно-генетических основах формирования многих видов психоневрологической патологии, традиционно уделяет эпилепсии немало внимания. Несмотря на это обстоятельство, в повседневной практике у детей и подростков нередко игнорируются нейрогенетические, иммунологические и аутоиммунные аспекты патогенеза эпилепсий, проявления психических нарушений и состояние их когнитивных функций, побочные эффекты антиэпилептических препаратов, альтернативные и дополнительные методы терапии и некоторые другие аспекты болезни.
В XV в. в книге «Молот ведьм» J. Sprenger и H. Kramer-Instintoris объясняли этиологию эпилепсии тем, что «… демоны с помощью чародейств телесно берут человека в обладание…». Хотя в настоящее время детские неврологи располагают современными методами лабораторно-инструментальной диагностики, а арсенал антиэпилептических лекарственных средств насчитывает уже десятки препаратов и продолжает пополняться, эпилепсия остается самым изучаемым и менее всего изученным состоянием.
Классификация эпилепсии
Окончательной и общепринятой классификации эпилепсии никогда не существовало, хотя с середины XX в. было предложено немало ее разновидностей.
- Penfield (1948) подразделял эпилепсию по причинам ее возникновения и морфологическим изменениям со стороны головного мозга, включая в свой вариант классификации эпилептических синдромов при различных активных церебральных процессах. Варианты классификации эпилепсий в свое время представили R. L. Masland (1960), R. N. Dejong (1964) и D. Janz (1969); последний в основу классификации поместил преобладание в клинической картине эпилепсии тех или иных видов приступов. Классификация эпилепсии по Z. Servit (1960) основывалась на патогенетическом принципе (предполагалось, что развитие церебральных пароксизмов обусловлено взаимодействием эпилептогенного раздражителя, эпилептогенного очага и судорожной готовности мозга).
Венская классификация (1964), принятая Международной противоэпилептической лигой (International League Against Epilepsy, ILAE), основывалась на клинико-феноменологическом и неврологическом (локализация патологического очага) принципах, а классификация по Сараджишвили П. М. (1969) явилась ее продолжением.
Классификация эпилептических приступов Международной противоэпилептической лиги образца 1969 г. была основана на достаточно большом (6) числе критериев:
1) клинический тип приступов;
2) тип приступов по электроэнцефалограмме (ЭЭГ);
3) интериктальная ЭЭГ-выраженность;
4) анатомический субстрат болезни;
5) этиология;
6) возраст пациентов.
При этом выделялись 4 больших группы припадков: парциальные приступы, генерализованные приступы, односторонние (или преимущественно односторонние) приступы, в также неклассифициируемые эпилептические пароксизмы. Парциальными считались приступы, при которых первые клинические симптомы болезни указывали на активацию анатомических и/или функциональных систем нейронов, ограниченную участком или одним полушарием мозга с соответствующей локализацией ЭЭГ-разряда. Парциальные припадки подразделялись на две подгруппы: с простой и сложной симптоматикой. Понятие генерализованных припадков предусматривало вовлечение в эпилептический процесс обоих полушарий мозга; в нескольких подгруппах рассматривались их судорожные или бессудорожные проявления.
В настоящее время разновидности эпилептических приступов у детей и взрослых рассматриваются в соответствии с классификацией, принятой в 1981 году в г. Киото (Япония). По типу имеющихся приступов предлагается выделять следующие их разновидности:
- Парциальные (фокальные, локальные) припадки: A — простые парциальные, B — сложные парциальные, C — парциальные с вторичной генерализацией.
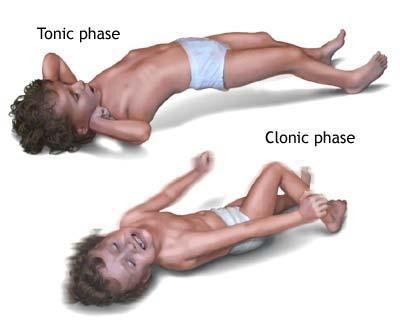
- Генерализованные припадки: A — абсансы (типичные и атипичные), B — миоклонические припадки, C — клонические припадки, D — тонические припадки, E — тонико-клонические припадки, F — атонические припадки.
- Неклассифицируемые эпилептические припадки.
В 1990-х гг. наибольшее распространение во всем мире получила классификация Международной противоэпилептической лиги, построенная на двух основных принципах дифференциации эпилепсии: разделение болезни на генерализованные и локализационно-обусловленные формы (по локализации эпилептических очагов), выделение эпилепсии известной этиологии (симптоматической) и неуточненной/неизвестной этиологии (идиопатической, криптогенной).
В классификации эпилепсий, эпилептических синдромов и схожих заболеваний, принятой Международной противоэпилептической лигой (Нью-Дели, 1989) выделяются следующие рубрики:
- Локализационно-обусловленные формы (очаговые, фокальные, локальные, парциальные):
1.1. Идиопатические (с возрастзависимым началом): доброкачественная эпилепсия детского возраста с центрально-височными пиками (роландическая); эпилепсия детского возраста с затылочными пароксизмами; первичная эпилепсия чтения.
1.2. Симптоматические: хроническая прогрессирующая парциальная эпилепсия Кожевникова; приступы, характеризующиеся специфическими способами провокации; другие формы эпилепсии с известной этиологией или органическими изменениями в мозге (лобная, височная, теменная, затылочная).
1.3. Криптогенные.
- Генерализованные формы эпилепсии:
2.1. Идиопатические (с возрастзависимым началом): доброкачественные семейные судороги новорожденных; доброкачественные судороги новорожденных; доброкачественная миоклоническая эпилепсия младенческого возраста; абсансная эпилепсия детская (пикнолепсия); абсансная эпилепсия юношеская; юношеская миоклоническая эпилепсия; эпилепсия с генерализованными судорожными приступами пробуждения; другие идиопатические генерализованные формы эпилепсии, не названные выше; формы, характеризующиеся специфическими способами провокации (чаще — фотосенситивная эпилепсия).
2.2. Криптогенные и/или симптоматические: синдром Веста (инфантильные спазмы); синдром Леннокса–Гасто; эпилепсия с миоклонически-астатическими приступами; эпилепсия с миоклоническими абсансами.
2.3. Симптоматические:
2.3.1. Неспецифической этиологии: ранняя миоклоническая энцефалопатия; ранняя младенческая эпилептическая энцефалопатия Ohtahara с паттерном «вспышка/угнетение» на ЭЭГ; другие симптоматические генерализованные формы эпилепсии, не названные выше.
2.3.2. Специфические синдромы.
- Эпилепсии, не имеющие четкой классификации как парциальные или генерализованные:
3.1. Имеющие как генерализованные, так и парциальные проявления: судороги новорожденных; тяжелая миоклоническая эпилепсия раннего детского возраста; эпилепсия с непрерывными пик-волнами во время медленного сна; приобретенная эпилептическая афазия Ландау–Клеффнера; другие неклассифицируемые формы эпилепсии, не определенные выше.
3.2. Приступы, не имеющие четких генерализованных или парциальных признаков.
- Специфические синдромы:
4.1. Ситуационно обусловленные приступы: фебрильные судороги; приступы, возникающие только по причине острых метаболических или токсических нарушений.
4.2. Изолированные приступы или изолированный эпилептический статус
4.3. Приступы, связанные исключительно с острым воздействием метаболических или токсических факторов, а также депривацией сна, приемом алкоголя и/или лекарственных средств, эклампсией и т. д.
Впоследствии было высказано мнение о необходимости внесения изменений в Международную классификацию эпилепсий и эпилептических синдромов, в связи с чем появились некоторые отличия в терминологии (по сравнению с классификацией Международной противоэпилептической лиги, 1989). «Криптогенные» формы эпилепсии было предложено заменить на «вероятно симптоматические», «парциальные» (приступы и формы эпилепсии) — на «фокальные», а слово «судороги» — на «приступы». Исключено подразделение парциальных (фокальных) приступов на простые и сложные (в зависимости от нарушения уровня сознания). Необходимо также отметить предложение по разделению эпилептических приступов на самокупирующиеся (генерализованные и фокальные) и продолжающиеся.
Самокупирующиеся приступы бывают следующими: генерализованные — тонико-клонические (включая варианты начала с клонической или миоклонической фазы), клонические (с легким тоническим компонентом или без него), типичные абсансы, атипичные абсансы, миоклонические абсансы, тонические, эпилептические спазмы, эпилептический миоклонус, массивный двухсторонний миоклонус, миоклонус век (с абсансами или без них), миоклонически-атонические (миатонические), негативный миоклонус, атонические, рефлекторные генерализованные; фокальные — сенсорные (с простыми симптомами, связанными с раздражением затылочной или теменной доли, или со сложными симптомами, связанными с раздражением височно-теменно-затылочной коры), моторные: клонические, асимметричные тонические (связанные с раздражением дополнительной моторной зоны — с типичными автоматизмами, с гиперкинетическими автоматизмами, с фокальным негативным миоклонусом, ингибиторные); геластические — гемиклонические, вторично-генерализованные, рефлекторные фокальные.
К продолжающимся приступам относят следующие: генерализованный эпилептический статус — статус генерализованных тонико-клонических приступов, статус клонических приступов, статус абсансов, статус тонических приступов, статус миоклонических приступов; фокальный эпилептический статус — кожевниковская эпилепсия, продолженная аура, статус лимбических приступов (психомоторный статус), гемиконвульсивный статус с гемипарезом.
Обновленная классификация эпилептических синдромов (ILAE, 2001) предусматривает выделение идиопатических фокальных эпилепсий младенчества и детства (доброкачественные младенческие приступы — несемейные, доброкачественная эпилепсия детства с центрально-височными спайками — роландическая, доброкачественная затылочная эпилепсия детства с ранним дебютом — тип Панайотопулоса, доброкачественная затылочная эпилепсия детства с поздним дебютом — тип Гасто), семейных (аутосомно-доминантных) фокальных эпилепсий (доброкачественные семейные приступы новорожденных, доброкачественные семейные приступы младенчества, аутосомно-доминантная ночная лобная эпилепсия, семейная височная эпилепсия, семейная фокальная эпилепсия с вариабельным фокусом), cимптоматических (или вероятно симптоматических) фокальных эпилепсий (лимбические — мезиальная височная эпилепсия с гиппокампальным склерозом, мезиальная височная эпилепсия вследствие специфической этиологии, другие формы определенной локализации и этиологии, неокортикальные эпилепсии — синдром Расмуссена, синдром гемиконвульсий-гемиплегии, мигрирующие парциальные приступы раннего младенчества, другие формы определенной локализации и этиологии); идиопатических генерализованных эпилепсий (доброкачественная миоклоническая эпилепсия младенчества, эпилепсия с миоклонически-астатическими приступами, детская абсансная эпилепсия, эпилепсия с миоклоническими абсансами); идиопатических генерализованных эпилепсий с вариабельным фенотипом (юношеская абсансная эпилепсия, юношеская миоклоническая эпилепсия, эпилепсия с изолированными генерализованными тонико-клоническими приступами, генерализованная эпилепсия с фебрильными приступами плюс); рефлекторных эпилепсий (идиопатическая фотосенситивная затылочная эпилепсия, другие формы эпилепсии с приступами вследствие зрительной стимуляции, первичная эпилепсия чтения, стартл-эпилепсия); эпилептических энцефалопатий (ранняя миоклоническая энцефалопатия, синдром Отахары, синдром Веста, синдром Драве, миоклонический статус при непрогрессирующих энцефалопатиях, синдром Леннокса–Гасто, синдром Ландау–Клеффнера, эпилепсия с продолженной спайк-волновой активностью во время медленного сна); прогрессирующих миоклонус-эпилепсий (специфические заболевания — болезнь Лафора, синдром Унферрихта–Лундборга, нейрональный цероидный липофусциноз и др.); приступов, для обозначения которых дефиниция «эпилепсия» необязательна (доброкачественные приступы новорожденных, фебрильные приступы, рефлекторные приступы; приступы, связанные с отменой алкоголя; приступы, вызванные лекарственными препаратами или другими химическими агентами; приступы, возникающие сразу после или в раннем периоде черепно-мозговой травмы, единичные приступы или единичные серии приступов, редко повторяемые приступы — олигоэпилепсия).
В МКБ-10 эпилепсии и эпилептическому статусу среди рубрик G40-G47 («Эпизодические и пароксизмальные расстройства») соответствуют рубрики G40 и G41: G40 — эпилепсия, G40.0 — локализованная (фокальная, парциальная) идиопатическая эпилепсия и эпилептические синдромы с судорожными припадками, G40.1 — локализованная (фокальная, парциальная) симптоматическая эпилепсия и эпилептические синдромы с простыми парциальными припадками, G40.2 — локализованная (фокальная, парциальная) симптоматическая эпилепсия и эпилептические синдромы с комплексными парциальными судорожными припадками, G40.3 — генерализованная идиопатическая эпилепсия и эпилептические синдромы, G40.4 — другие виды генерализованной эпилепсии и эпилептических синдромов, G40.5 — особые эпилептические синдромы, G40.6 — припадки grand mal неуточненные (с малыми припадками (petit mal) или без них), G40.7 — малые припадки (petit mal) неуточненные без припадков grand mal, G40.8 — другие уточненные формы эпилепсии, G40.9 — эпилепсия неуточненная, G41 — эпилептический статус, G41.0 — эпилептический статус grand mal (судорожных припадков), G41.1 — эпилептический статус petit mal (малых припадков), G41.2 — сложный парциальный эпилептический статус, G41.8 — другой уточненный эпилептический статус, G41.9 — эпилептический статус неуточненный.
- I. Wolf и соавт. (2005) предлагают соотносить основные виды метаболических эпилепсий с патогенетическими факторами следующим образом: 1) дефицит энергии: гипогликемия, дефицит транспортера глюкозы-1, недостаточность дыхательной цепи, дефицит креатина; 2) токсический эффект: аминоацидопатии, органические ацидурии, дефекты цикла обмена мочевины; 3) нарушения нейрональной функции: болезни накопления; 4) расстройства систем нейротрансмиттеров: некетотическая гиперглицинемия, дефицит ГАМК-трансаминазы (ГАМК — гамма-аминомасляная кислота), недостаточность янтарной полуальдегид дегидрогеназы; 5) ассоциированные церебральные мальформации: пероксисомные нарушения (синдром Цельвегера), дефекты О-гликозилирования; 6) зависимость от витаминов/кофакторов: биотинидазная недостаточность, витамин В6-зависимая эпилепсия, пиридоксальфосфат-зависимая эпилепсия, судороги, зависимые от фолиевой кислоты; болезнь Менкеса (болезнь мочи с запахом кленового сиропа); 7) прочие (разные) метаболические причины: врожденные дефекты гликозилирования, дефицит биосинтеза серина, врожденные ошибки церебральной возбудимости (нарушения со стороны ионных каналов).
Этиология и патогенез
Механизмы эпилептогенеза в детском возрасте имеют дополнительные особенности. В результате разбалансировки между торможением и возбуждением незрелый мозг ребенка чаще реагирует развитием эпилептических приступов.
Этиологические факторы эпилепсии включают острую и хроническую интоксикацию, травмы и инфекционные заболевания центральной нервной системы (ЦНС), генетическую и метаболическую предрасположенность, гипоксию, ишемию и т. д. Эпилептические приступы нередко сопровождают врожденные нарушения формирования ЦНС (артериовенозную фистулу, врожденную гидроцефалию, порэнцефалию, микрогирию, агенезию мозолистого тела, гидроанэнцефалию и другие виды дизгенезий головного мозга), опухоли головного мозга, органические ацидемии, а также травмы головного мозга и черепно-мозговые травмы и т. д.
Среди эпилепсий, ассоциированных с мальформациями мозга, основной патологией следует считать тяжелую форму синдрома Цельвегера, для которой характерны мальформации развития коркового вещества мозга (полимикрогирия фронтального и оперкулярного регионов, пахигирия). Принято считать, что некоторые формы нарушений 0-гликозилирования (синдром Уокера–Варбурга, болезнь «мышц/глаз/мозга», мышечная дистрофия типа Фукуяма) приводят к мальформациям головного мозга в виде «булыжной» лиссэнцефалии с сопутствующими фармакорезистентными эпилептическими приступами.
Основными патогенетическими механизмами эпилепсии принято считать аномальное возбуждение и торможение нейрональных мембран и нейронов нейромедиаторами и распространение судорожной активности. Существует также ряд иммунологических, нейрохимических, метаболических и генетических факторов, имеющих значение в патогенезе формирования эпилепсии в детском возрасте.
На клеточном уровне при эпилепсии происходят многочисленные изменения в функциях ионных каналов, регуляции рецепторов нейротрансмиттеров и метаболизма энергии, приводящие к развитию приступов. Кора головного мозга человека состоит из мелких клеточных сетей, функции которых легко нарушаются, приводя к развитию гиперсинхронных разрядов.
При эпилепсии нейроны фокального региона или всего головного мозга синхронно активируются (в норме они относительно десинхронизированы). Под воздействием различных раздражающих факторов нормальный уровень нейрональной активации трансформируется в синхронные разряды, которые могут вовлекать нервные клетки в паттерн аномальной активации. Тип эпилептического приступа при этом зависит от количества и локализации клеток ЦНС, задействованных в процессе патологической активации.
Анатомическая микроструктура коры головного мозга человека и ее физиологические особенности являются достаточным субстратом для развития и распространения гиперсинхронной активности. Если для развития единичного эпизода судорожной активности требуется лишь кратковременное нарушение физиологии ЦНС, то повторные и непровоцируемые эпилептические припадки обычно сопряжены уже с выраженными анатомическими и патофизиологическими нарушениями, а также с генетическими дефектами. Есть мнение, что большинство форм эпилепсии с первичной генерализацией генетически детерминированы.
Установлена или предполагается генетическая природа многих форм эпилепсии, а также заболеваний, не являющихся эпилептическими синдромами, но в ряде случаев протекающих со специфическими приступами того или иного типа.
Одним из специфических синдромов эпилепсии с генетической детеминировнностью является синдром Веста (OMIM #308350, генный локус Xp22.13); существуют еще 3 типа ранней инфантильной эпилептической энцефалопатии, подпадающие под понятие «инфантильные спазмы»: EIEE2 (OMIM 300672), EIEE3 (OMIM 609304) и EIEE4 (OMIM 612164).
Другими наследственно-обусловленными разновидностями эпилепсии являются следующие: синдром Леннокса–Гасто (OMIM #606369, генный локус 4q21.3), синдром Ландау–Клеффнера (OMIM 245570), синдром Гасто (OMIM 132090), синдром Драве (OMIM #607208), синдром Отахары (OMIM #308350, генный локус Xp22.13) и др.
Определено около 100 генов, ответственных за возникновение различных форм эпилепсии. В международной онлайн-базе данных «Менделевское наследование у человека» (On-line Mendelian Inheritance in Men, OMIM) представлено немало разновидностей эпилепсии и генетических синдромов, протекающих с проявлениями эпилепсии, например: синдром Айкарди (OMIM 304050), болезнь Александера (OMIM #203450, генные локусы 17q21, 11q13), синдром Ангельмана (OMIM 105830), синдром Блоха–Сульцбергера (OMIM #308300), болезнь Лафора (OMIM #254780, генный локус 6q23–25), синдром Миллера–Дикера (OMIM #247200, генный локус 17p13.3),), синдром Смит–Лемли–Опитца (OMIM #270400, генный локус 11q12-q13), синдром Sturge–Weber (OMIM 185300), синдром Унферрихта–Лундборга (OMIM #254800), туберозный склероз (OMIM #191100), изовалериановая ацидемия (OMIM 243500), метилмалоновая ацидемия (различные типы по OMIM 251100, 251110, 277400, 277410, 277380, 606169, 251000, генетический локус 6p21.2-p12), пропионовая ацидемия (OMIM #606054), галактосиалидоз — cиндром Голдберга (OMIM 256540, генный локус 20q13.1), болезнь Гоше трех типов (OMIM #230800, 1q21; #230900, 1q21; #231000, 1q21), синдром Leigh (OMIM #256000, генные локусы 11q13, 11p11.11, 9q34, 8q22.1, 7q31-q32, 5q31.2, 5q11.1, 5p15, 2q33, 19p13), нейрональный цероидный липофусциноз (OMIM 600143) и его варианты — ранний инфантильный тип 1 (OMIM #256730, генный локус 1р32), поздний инфантильный, тип 2 (OMIM #204500, генный локус 11р15.5), ювенильный вариант, тип 3 (OMIM #610003, генный локус 8p23), семейные фебрильные судороги тип 1 (OMIM 121210, генный локус 8q13-q21), тип 2 (OMIM 602477, генный локус 19p), тип 3 (OMIM 604403, генный локус 2q24), тип 4 (OMIM 604352, генный локус 5q14), тип 5 (OMIM 609255, генный локус 6q), тип 6 (OMIM 609253, генный локус 18p), тип 7 (OMIM 611515, генный локус 21q22), тип 8 (OMIM 611277, генный локус 5q31), тип 9 (OMIM 611634, генный локус 3p24.2-p23), тип 10 (OMIM 612637, генный локус 3q26), болезнь мочи с запахом кленового сиропа (OMIM #309400), нейрофиброматоз тип 1 (OMIM 162200, генный локус 17q11.2) и тип 2 (OMIM #101000, генный локус 22q12.2), синдром Ретта (OMIM #312750 и #300672) и др.
Выделяют несколько механизмов аномальной активации нейронов; происходит усиление местных эксайтоторных цепей (хроническое — вследствие реорганизации синаптических цепей при повреждении головного мозга, острое — в результате нарастания синаптической эффективности при высокой активации нейронов). Последнее возможно при задействовании рецепторов N-метил-D-аспартата (NMDA). Формируется система положительной обратной связи между сетью и самой природой эксайтоторной синаптической активности.
Кроме того, нейроны могут синхронизироваться окружающими их внеклеточными потоками (в ограниченном внеклеточном пространстве), что происходит под воздействием повышения внеклеточного калия и снижения внеклеточного кальция, а также электрического взаимодействия между некоторыми клетками. Все эти факторы в норме присутствуют в клетках коры головного мозга, но при нормальной активности ЦНС они активируются на сравнительно низком уровне (избыточная активация предотвращается мощными ингибиторными механизмами). Описываемая ингибиция носит как синаптический характер (с задействованием рецепторов ГАМК типов А и В — ГАМКА и ГАМКВ), так и объясняется активацией внутренних вольтаж-зависимых потоков калия.
Если по какой-либо причине происходит снижение уровня ингибиции, препятствующего локальной гиперсинхронизированной активности, то формируется паттерн регенеративной активности (приводя к фокальному эпилептическому разряду). Дальнейшая поломка механизмов ингибиторного контроля сопровождается распространением возбуждающей активность на близко и далеко расположенные отделы мозга с развитием манифестного эпилептического приступа.
Эпилепсия традиционно рассматривается в качестве одной из моделей иммунной патологии, что связано с высокой вероятностью участия различных иммунологических реакций в патогенезе многих форм эпилепсии.
Гипотеза об участии иммунных механизмов в патогенезе эпилепсии была высказана A. E. Walker (1969), но отечественные исследователи А. Д. Адо и Т. М. Царегородцева (1968) описали механизмы аллергических реакций при аутоиммунных поражениях нервной системы еще до него. В середине 1980-х гг. были выявлены аномалии соотношения субпопуляций CD4/CD8 (хелперов и супрессоров) у пациентов с эпилепсией, а в начале 1990-х гг. появились сообщения об изменениях соотношения каппа/лямбда-легких цепей иммуноглобулинов у детей с резистентными формами эпилепсии.
Иммуногенетические исследования при эпилепсии были преимущественно сосредоточены на изучении системы HLA; в частности, было обнаружено повышение встречаемости аллели HLA-DRW13 (W6) у пациентов с ювенильной миоклонус-эпилепсией (39,5%), повышение встречаемости HLA-DR5 и снижение HLA-DR4 при криптогенном синдроме Леннокса–Гасто (55%), повышение встречаемости HLA-A2 у пациентов с семейными случаями генерализовaнной формы эпилепсии (89%).
Отечественными исследователями отмечены изменения в структуре популяций иммунокомпетентных клеток, характеризующих систему цитокинов (повышение по маркерам CD25 — низкоаффинная субъединица рецептора интерлейкина 2 (ИЛ-2), CD122 — высокоаффинная субъединица рецептора ИЛ-2), то есть признаки аутоиммунного компонента эпилептического процесса. Зарубежными исследователями у детей с резистентными формами эпилепсии (синдром Веста, Леннокса–Гасто) в крови обнаруживались даже кортикальные тимоциты (CD1+).
Продолжает оставаться не выясненным клиническое значение нейросенсибилизации лимфоцитов при эпилепсии в детском возрасте. Известно, что при эпилепсии в ЦНС индуцируется синтез аутоантител различной нейроспецифичности, а их потенциальный вклад в патогенетический механизм заболевания считается чрезвычайно значимым у детей, хотя речь может идти не обо всех формах эпилепсии.
При эпилепсии патогенетическое значение могут иметь нейроиммунные процессы, вызываемые антителами к специфическим сайтам глутаматных и холиновых рецепторов, ганглиозидам, а также нейрональным элементам эпилептических очагов (кора, гиппокамп, амигдала, наружное коленчатое тело).
Имеются публикации, указывающие на роль аллергических и/или аутоиммунных реакций в патогенезе эпилепсии в детском возрасте (в частности, провокация эпилепсии пищевой непереносимостью или аллергией, инфантильных спазмов — вакцинацией АКДС и т. д.). По мнению J. Choi и S. Koh (2008), воспаление играет немаловажную роль в процессах иммунного эпилептогенеза (воспалительные и иммунные реакции оказывают несомненное влияние на возбудимость нейронов). Именно хроническое церебральное воспаление в ряде случаев определяет фармакорезистентность эпилептических приступов.
Каждый нейрон, находящийся в эпилептогенном очаге, обладает способностью к стереотипическому и синхронизированному электроответу (пароксизмальное деполяризующее переключение). В качестве эпилептического очага рассматриваются несколько аномально функционирующих нейронов, действующих патологически синхронно. Общепринятым критерием эпилептизации нейрона является деполяризация мембраны (уменьшение величины отрицательного заряда при поляризации). Возникновение эпилептического приступа — результат интрацеллюлярной пароксизмальной деполяризации, которая в интериктальном периоде сменяется глобальной деполяризацией.
Формирование симптоматической эпилепсии у детей с детским церебральным параличом и гидроцефалией является вполне ожидаемым явлением, как и исход перинатального поражения нервной системы. Анатомо-морфологические изменения головного мозга при этих заболеваниях ЦНС являются органическим субстратом для возникновения аномальной электрической активности с генерацией эпилептических пароксизмов. При фебрильных судорогах трансформация имеющихся пароксизмов в симптоматическую эпилепсию происходит в ходе преобразования фебрильных судорожных эпизодов в афебрильные, чему способствует относительная незрелость церебральных структур у детей грудного, раннего и дошкольного возраста.
Существует немало эпилепсий метаболического происхождения. Некоторые из них ассоциированы с нарушениями обмена креатина (Х-связанный дефект транспортера креатина, нарушения синтеза креатина при гуанидиноацетат метилтрансферазной недостаточности, нарушения синтеза креатина при аргинин-глицин амидинотрансферазной недостаточности), нарушениями нейрональных функций вследствие болезней накопления (болезнь Тей-Сакса, сиалидоз тип I и II, нейрональный цероидный липофусциноз — типы I, II, III и IV), нарушениями цикла мочевины (гиперорнитинемия-гипераммониемия-гомоцитруллинурия, гипераргининемия и др.), нарушениями метаболизма аминокислот (фенилкетонурия, лейциноз и др.), нарушениями метаболизма органических кислот (метилмалоновая и пропионовая ацидемия, глутаровая ацидурия тип I и др.), нарушениями метаболизма пурина и пиримидина (дефицит аденилосукцинатлиазы и др.), нарушениями нейротрансмиттерных систем (некетотическая гиперглицинемия и др.), нарушениями метаболизма ГАМК (недостаточность янтарной полуальдегиддегидрогеназы).
В группу витаминозависимых эпилепсий, помимо витамин В6-зависимой эпилепсии, входят пиридоксаминфосфатоксигеназная недостаточность, фолатзависимые судороги, дефицит биотинидазы и голокарбоксилазосинтазы.
Хотя у многих детей с митохондриальной патологией не отмечается эпилепсии, соответствующие приступы считаются характерными проявлениями митохондриальных цитопатий. N. I. Wolf и соавт. (2005) подчеркивают, что в младенческом и детском возрасте эпилепсия сопутствует 26–60% митохондриальных нарушений. В классическом варианте эпилепсия обычно сопутствует синдромам MERRF (Myoclonic Epilepsy with Ragged Red Fibres — миоклоническая эпилепсия с рваными мышечными волокнами) и MELAS (Mitochondrial Encephalomyopathy, Lactic Acidosis, and Stroke-like episodes — митохондриальная энцефаломиопатия, лактатацидоз, инсультоподобные эпизоды). При митохондриальной патологии наиболее часто встречаются две разновидности эпилептических синдромов: прогрессирующая миоклонус-эпилепсия и синдром Веста.
К числу факторов, провоцирующих рефлекторные приступы при эпилепсии, относятся зрительные стимулы (мелькающий свет, фотосенситивный паттерн, другие зрительные стимулы), мыслительный процесс, музыка, еда, выполнение движений, соматосенсорные стимулы, проприоцептивные стимулы, чтение, горячая вода, резкие звуки.
Предполагается, что в будущем лишь комплексные исследования (нейрогенетические, нейроиммунологические, нейрохимические) позволят получить объективную информацию о процессах, происходящих в ЦНС при эпилепсии у детей, уточнив патогенетические механизмы этой группы хронических пароксизмальных церебральных нарушений.
Литература
- Броун Т. Р., Холмс Г. Л. Эпилепсия. Клиническое руководство. Пер. с англ. М.: «Изд-во БИНОМ». 2006. 288 с.
- Киссин М. Я. Клиническая эпилептология. М.: ГЭОТАР-Медиа. 2009. 256 с.
- Студеникин В. М., Шелковский В. И., Балканская С. В. Локализационно-обусловленные формы эпилепсии у детей и их лечение // Лечащий Врач. 2008. № 5. С. 68–71.
- Эпилепсия в нейропедиатрии (коллективная монография) / Под ред. Студеникина В. М. М.: Династия. 2011, 440 с.
- Arzimanoglou A., Guerrini R., Aicardi J. Aicardi’s epilepsy in children. 3 rd ed. Philadelphia-Tokyo. Wolters Kluwer. 2004. 516 p.
- Berg A. T., Blackstone N. W. Concepts in classification and their relevance to epilepsy // Epilepsy Res. 2006. 70. Suppl. S. 11–19.
- Chapman K., Rho J. M. Pediatric epilepsy case studies. From infancy and childhood through infancy. CRC Press/Taylor&Francis Group. Boca Raton–London. 2009. 294 p.
- Choi J., Koch S. Role of brain inflammation in epileptogenesis // Yonsei Med. J. 2008. 49. P. 1–18.
- Schwartzkroin P. ed. Encyclopedia of basic epilepsy research/Three-volume set. 1–3. Philadelphia. Elsevier/Academic Press. 2009. 2496 p.
- Engel J., Pedley T. A. eds. Epilepsy: A comprehensive textbook. 2 nd ed. 1–4. Lippincott Williams&Wilkins/A Wolters Kluwer Business. 2008. 2986 p.
- Roger J., Bureau M., Dravet Ch., Genton P. et al. eds. Epileptic syndromes in infancy, childhood and adolescence. 4 th ed. (with video). Montrouge (France). John Libbey Eurotext. 2005. 604 p.
- Goodman M., Lamm S. H., Bellaman M. H. Temporal relationship modeling: DTP or DT immunization and infantile spasms // Vaccine. Vol. 16. P. 225–231.
- Levite M. Autoimmune epilepsy // Nat. 2002. Vol. 3. P. 500.
- Montelli T. C., Soares A. M., Peracoli M. T. Immunologic aspects of West syndrome and evidence of plasma inhibitory effects on T cell function // Arq. 2003. Vol. 61. P. 731–737.
- Palace J., Lang B. Epilepsy: an autoimmune disease? // J. Neurol. Psychiatry. 2000. Vol. 69. P. 711–714.
- Rosenberg R. N., DiMauro S., Paulson H. L., Ptack L., Nestler E. J. (eds.) The molecular and genetic basis of neurologic and psychiatric disease. 4 th ed. Philadelphia-Baltimore. Lippincott Williams&Wilkins/A Wolters Kluwer Business. 882 p.
- Soltesz I., Staley K. (eds). Computational neuroscience in epilepsy. Amsterdam-Boston. Academic Press/An Imprint of Elsevier. 2008. 604 p.
- Specchio N., Fusco L., Claps D., Vigevano F. Epileptic encephalopathy in children possibly related to immune-mediated pathogenesis // Brain Dev. 2010. 32. P. 51–56.
- Wallace S. J., Farrell K. (eds.). Epilepsy in children. London. Arnold Press. 497 p.
- Wolf N. I., Bast T., Surtees R. Epilepsy in inborn errors of metabolism // Epileptic Disord. Vol. 7. P. 67–81.
Источник: Журнал «Лечащий врач» №6, 2014
Эпилепсия проявляется повторными непровоцированными приступами, носящими характер разнообразных внезапных и транзиторных патологических феноменов, затрагивающих сознание, двигательную и чувствительную сферы, вегетативную нервную систему, а также психику пациента. Два приступа, возникшие у пациента в течение 24 часов, считаются единым событием [1].
Клинические проявления эпилепсии вариабельны и многообразны. Они преимущественно зависят как от формы болезни, так и от возраста пациентов. Возрастные аспекты эпилепсии в детской неврологии предполагают необходимость четкого выделения возрастзависимых форм этой группы болезней [1, 2].
Клинические проявления эпилепсии у детей и подростков
Клиническая картина эпилепсии включает два периода: приступный и межприступный (интериктальный). Проявления болезни определяются имеющимся у пациента типом приступов и локализацией эпилептогенного очага. В интериктальном периоде неврологическая симптоматика у больного может полностью отсутствовать. В других случаях неврологические симптомы у детей могут быть обусловлены заболеванием, вызывающим эпилепсию [1, 3].
Парциальные приступы
Проявления простых парциальных приступов зависят от места локализации эпилептогенного очага (лобная, височная, теменная, затылочная доли, перироландическая область и т. д.). До 60–80% эпилептических приступов у детей и совершеннолетних пациентов относятся к числу парциальных. Эти приступы протекают у детей с различными феноменами: моторными (тонические или клонические судороги в верхних или нижних конечностях, на лице — контралатерально имеющемуся очагу), соматосенсорными (ощущение онемения или «прохождения тока» в конечностях или половине лица, противоположных эпилептогенному очагу), специфическими сенсорными (простые галлюцинации — акустические и/или зрительные), вегетативными (мидриаз, потоотделение, бледность или гиперемия кожных покровов, неприятные ощущения в эпигастральной области и др.) и психическими (преходящие нарушения речи и др.) [1, 4].
Клинические проявления парциальных приступов являются маркерами топики эпилептического очага. При локализации очагов в двигательной коре приступы, как правило, характеризуются фокальными тоническими и клоническими судорогами — моторные припадки джексоновского типа. Сенсорные джексоновские припадки (фокальные парестезии) возникают при наличии эпилептического очага в задней центральной извилине. Зрительные припадки (простые парциальные), характеризующиеся соответствующими феноменами (искры света, зигзаги перед глазами и т. д.), возникают при расположении эпилептических очагов в затылочной коре. Различные обонятельные (неприятный запах), акустические (ощущение шума в ушах) или вкусовые (неприятный вкус) феномены возникают при локализации очагов, соответственно, в области обонятельной, слуховой или вкусовой коры. Очаги, расположенные в премоторной коре, индуцируют адверсивные припадки (сочетание отведения глазных яблок и головы с последующими клоническими подергиваниями); нередко такие приступы трансформируются во вторично-генерализованные. Парциальные приступы бывают простыми и сложными [1, 3].
Простые парциальные приступы (ППП). Проявления зависят от локализации эпилептического очага (локализационно-обусловленные). ППП являются моторными и протекают без изменения или потери сознания, поэтому ребенок в состоянии рассказать о своих ощущениях (кроме тех случаев, когда приступы происходят во время сна) [1].
Для ППП характерно возникновение судорог в одной из верхних конечностей или в области лица. Эти приступы приводят к отклонению головы и отведению глаз в сторону полушария, контралатерального локализации эпилептического очага. Фокальные судороги могут начаться на ограниченном участке или генерализоваться, напоминая вторично-генерализованные тонико-клонические судороги. Паралич (или парез) Тодда, выражающийся в транзиторной слабости в течение от нескольких минут до нескольких часов, а также отведение глазных яблок в сторону пораженного полушария служат указаниями на эпилептогенный очаг. Указанные феномены появляются у пациента после ППП (постиктальный период) [1, 4].
Простые парциальные вегетативные приступы (ППВП). Предлагается отдельно выделять эту разновидность сравнительно редко встречающихся эпилептических припадков. ППВП индуцируются эпилептогенными очагами, локализованными в орбито-инсуло-темпоральной области. При ППВП преобладают вегетативные симптомы (потоотделение, внезапное сердцебиение, абдоминальный дискомфорт, урчание в животе и т. д.). Вегетативные проявления при эпилепсии довольно многообразны и определяются дигестивными, кардиальными, дыхательными, зрачковыми и некоторыми другими симптомами. Абдоминальные и эпигастральные эпилептические приступы считаются более характерными для детей в возрасте от 3 до 7 лет, а кардиальные и фарингооральные чаще встречаются в более старшем возрасте. Дыхательные и зрачковые ППВП свойственны для эпилепсии у пациентов любого возраста. Так, клинически абдоминальные эпилептические приступы обычно характеризуются возникновением резких болей в области живота (иногда в сочетании с рвотой). Эпигастральные ППВП проявляются в виде различных признаков дискомфорта в эпигастральной области (урчание в животе, тошнота, рвота и т. д.). Кардиальные эпилептические приступы манифестируют в виде тахикардии, повышения артериального давления, болей в области сердца («эпилептическая грудная жаба»). Фарингооральные ППВП — это эпилептические пароксизмы, выражающиеся в гиперсаливации, нередко в сочетании с движениями губ и/или языка, глотанием, облизыванием, пожевыванием и т. д. Основным проявлением зрачковых ППВП служит появление мидриаза (так называемая «зрачковая эпилепсия»). Респираторные ППВП характеризуются приступами нарушения дыхания — одышки («эпилептическая астма») [1].
Сложные парциальные приступы (СПП). Манифестация СПП весьма многообразна, но во всех случаях у пациентов отмечаются изменения сознания. Зафиксировать нарушения сознания у детей грудного и раннего возраста довольно сложно. Начало СПП может выражаться в виде простого парциального припадка (ППП) с последующим нарушением сознания; изменения сознания также могут возникать непосредственно в приступе. СПП часто (примерно в половине случаев) начинаются с эпилептической ауры (головная боль, головокружение, слабость, сонливость, неприятные ощущения в полости рта, тошнота, дискомфорт в области желудка, онемение губ, языка или рук; транзиторная афазия, ощущение сдавления в горле, затруднение дыхания, слуховые и/или обонятельные пароксизмы, необычное восприятие всего окружающего, ощущения déjà vu (уже пережитое) или jamais vu (впервые видимое, слышимое и никогда не переживаемое) и т. д.), позволяющей уточнить локализацию эпилептогенного очага. Такие феномены, как судорожные клонические движения, насильственная девиация головы и глаз, фокальное тоническое напряжение и/или разнообразные автоматизмы (повторяющаяся двигательная нецеленаправленная деятельность: облизывание губ, глотательные или жевательные движения, вычурные движения пальцев, рук и лицевой мускулатуры, у начавших ходить — бег и т. д.) могут сопутствовать СПП. Автоматизированные движения при СПП не являются целенаправленными; контакт с пациентом во время приступа утрачивается. В грудном и раннем возрасте описываемые автоматизмы обычно не выражены [1, 4].
Парциальные приступы с вторичной генерализацией (ППВГ). Вторично-генерализованные парциальные приступы бывают тоническими, клоническими или тонико-клоническими. ППВГ всегда протекают с потерей сознания. Могут возникать у детей и подростков как после простого, так и после сложного парциального приступа. У пациентов возможно наличие эпилептической ауры (около 75% случаев), предшествующей ППВГ. Аура обычно носит индивидуальный характер и бывает стереотипной, а в зависимости от поражения той или иной области головного мозга бывает моторной, сенсорной, вегетативной, психической или речевой [1, 4].
Во время ППВГ пациенты теряют сознание; они падают, если находятся не в лежачем положении. Падение обычно сопровождается специфическим громким криком, что объясняется спазмом голосовой щели и судорожным сокращением мускулатуры грудной клетки [1].
Генерализованные приступы (первично-генерализованные)
Как и парциальные (фокальные) эпилептические припадки, генерализованные приступы у детей достаточно разнообразны, хотя являются несколько более стереотипными.
Клонические приступы. Выражаются в виде клонических судорог, которые начинаются с внезапно возникающей гипотонии или короткого тонического спазма, за которыми следуют двухсторонние (но нередко асимметричные) подергивания, могущие преобладать в одной конечности [1, 4]. Во время приступа отмечаются различия в амплитуде и частоте описанных пароксизмальных движений. Клонические судороги обычно наблюдаются у новорожденных и детей грудного и раннего возраста.
Тонические приступы. Эти судорожные припадки выражаются в кратковременном сокращении мышц-экстензоров. Тонические приступы характерны для синдрома Леннокса–Гасто, они отмечаются и при других видах симптоматических эпилепсий. Тонические приступы у детей чаще происходят во время фазы не-REM-сна, чем при бодрствовании или в фазе REM-сна. При сопутствующем сокращении дыхательной мускулатуры тонические судороги могут сопровождаться развитием апноэ [1].
Тонико-клонические приступы (ТКП). Выражаются в виде судорог, протекающих по типу grand mal. Для ТКП характерна тоническая фаза продолжительностью менее 1 минуты, сопровождаемая заведением глаз вверх. Одновременно происходит снижение газообмена, обусловленное тоническим сокращением дыхательной мускулатуры, что сопровождается появлением цианоза. Клоническая судорожная фаза приступа следует за тонической и выражается в клоническом подергивании конечностей (обычно в течение 1–5 минут); газообмен при этом улучшается или нормализуется. ТКП могут сопровождаться гиперсаливацией, тахикардией, а также метаболическим и/или дыхательным ацидозом. При ТКП постиктальное состояние чаще продолжается менее 1 часа [1, 4].
Абсансные приступы (абсансы). Протекают по типу petit mal («малого эпилептического припадка») и представляют собой кратковременную потерю сознания с последующей амнезией («замирание»). Абсансам могут сопутствовать клонические подергивания век или конечностей, расширение зрачков (мидриаз), изменения мышечного тонуса и цвета кожных покровов, тахикардия, пилоэреция (сокращение мышц, поднимающих волосы) и различные двигательные автоматизмы [1, 3, 4].
Абсансы бывают простыми и сложными. Простые абсансы представляют собой приступы кратковременного отключения сознания (с характерными медленными волнами на ЭЭГ). Сложные абсансы — это нарушения сознания, сочетающиеся с атонией, автоматизмами, мышечным гипертонусом, миоклониями, приступами кашля или чихания, а также вазомоторными реакциями. Принято выделять также субклинические абсансы, то есть транзиторные нарушения без выраженных клинических проявлений, отмечаемые при нейропсихологическом обследовании и сопровождающиеся при ЭЭГ-исследовании медленно-волновой активностью [1].
Простые абсансы встречаются значительно реже, чем сложные. При наличии у пациента ауры, очаговой двигательной активности в конечностях и постиктальной слабости, замирания не расцениваются в качестве абсансов (в таких случаях следует думать о сложных парциальных приступах) [1].
Псевдоабсансы. Этот тип припадков описан H. Gastaut (1954) и трудноотличим от истинных абсансов. При псевдоабсансах также наблюдается кратковременное выключение сознания с остановкой взора, но начало и окончание припадка несколько замедлены. Сами псевдоабсансные приступы более продолжительны по времени и нередко сопровождаются парестезиями, феноменом déjà vu, выраженными вегетативными расстройствами, часто — постиктальной оглушенностью. Псевдоабсансы относятся к парциальным (фокальным) височным пароксизмам. Решающее значение в дифференциации псевдоабсансов от истинных абсансов имеет ЭЭГ-исследование [1, 3].
Миоклонические припадки (эпилептический миоклонус). Миоклонические подергивания бывают изолированными или рецидивирующими. Миоклонии характеризуются короткой продолжительностью и быстрыми двухсторонними симметричными мышечными сокращениями, а также вовлечением различных групп мышц. Миоклонии обычно отмечаются у детей с доброкачественными или симптоматическими формами эпилепсии. В структуре группы симптоматических эпилепсий миоклонии могут отмечаться как при различных непрогрессирующих формах болезни (синдром Леннокса–Гасто и др.), так и при сравнительно редких прогрессирующих формах миоклонус-эпилепсии (болезнь Лафора, болезнь Унферрихта–Лундборга, синдром MERRF, нейрональный цероидный липофусциноз и др.). Иногда миоклоническая активность ассоциируется с атоническими приступами; при этом дети могут падать при ходьбе [1, 4].
Атонические приступы. Характеризуются внезапным падением умеющего стоять и/или ходить ребенка, то есть отмечается так называемая «дроп-атака» (drop-attack). При атоническом приступе происходит внезапное и выраженное снижение тонуса в мышцах конечностей, шеи и туловища. Во время атонического приступа, началу которого может сопутствовать миоклония, сознание у ребенка нарушается. Атонические приступы чаще отмечаются у детей с симптоматическими генерализованными эпилепсиями, но при первично-генерализованных формах болезни встречаются сравнительно редко [1, 3, 4].
Акинетические приступы. Напоминают атонические припадки, но, в отличие от них, при акинетических приступах у ребенка отмечается внезапная обездвиженность без значительного снижения мышечного тонуса [1].
Термопатологические проявления эпилепсии
Еще в 1942 г. A. M. Hoffman и F. W. Pobirs высказали предположение, что приступы избыточного потоотделения являются одной из форм «фокальной автономной эпилепсии». H. Berger (1966) впервые описал лихорадку (гипертермию) в качестве необычного проявления эпилепсии, а впоследствии D. F. Сohn и соавт. (1984) подтвердили этот термопатологический феномен, назвав его «термальной эпилепсией» [1, 5]. О возможности манифестации интермиттирующей лихорадки или «фебрильных крампи» при эпилепсии сообщают S. Schmoigl и L. Hohenauer (1966), H. Doose и соавт. (1966, 1970), а также K. M. Chan (1992).
- J. Wachtel и соавт. (1987) считают, что генерализованные тонико-клонические приступы могут приводить к гипертермии; в их наблюдениях у 40 пациентов из 93 (43%) в момент приступа отмечался подъем температуры свыше 37,8 °С. J. D. Semel (1987) описал сложный парциальный эпилептический статус, манифестирующий в виде «лихорадки неизвестного происхождения» [1].
В ряде случаев эпилепсия может проявляться в виде гипотермии. R. H. Fox и соавт. (1973), D. J. Thomas и I. D. Green (1973) описали спонтанную периодическую гипотермию при диэнцефальной эпилепсии, а M. H. Johnson и S. N. Jones (1985) наблюдали эпилептический статус с гипотермией и метаболическими нарушениями у пациента с агенезией мозолистого тела. W. R. Shapiro и F. Blum (1969) описали спонтанную рецидивирующую гипотермию с гипергидрозом (синдром Шапиро). В классическом варианте синдром Шапиро представляет сочетание агенезии мозолистого тела с пароксизмальной гипотермией и гипергидрозом (холодный пот), а патогенетически связан с вовлечением в патологический процесс гипоталамуса и других структур лимбической системы. Разные исследователи называют синдром Шапиро «спонтанной периодической гипотермией» или «эпизодической спонтанной гипотермией». Представлено описание спонтанной периодической гипотермии и гипергидроза без агенезии corpus callosum. K. Hirayama и соавт. (1994), а затем K. L. Lin и H. S. Wang (2005) описали «реверсивный синдром Шапиро» (агенезия мозолистого тела с периодической гипертермией вместо гипотермии) [1, 6].
В большинстве случаев пароксизмальные гипотермии рассматриваются как связанные с диэнцефальной эпилепсией. Хотя, по мнению C. Bosacki и соавт. (2005), гипотеза «диэнцефальной эпилепсии» применительно к эпизодической гипотермии недостаточно убедительна, эпилептическое происхождение, по крайней мере, части случаев синдрома Шапиро и подобных ему состояний подтверждается тем, что антиэпилептические препараты предупреждают развитие приступов гипотермии и гипергидроза [1, 3].
Гипертермия или гипотермия не могут быть четко отнесены к фокальным или генерализованным пароксизмам, но саму возможность проявления эпилептических приступов у детей в виде выраженных температурных реакций (изолированно или в сочетании с другими патологическими феноменами) не следует игнорировать [1, 7].
Психические особенности детей с эпилепсией
Многие психические изменения у детей и подростков с эпилепсией остаются не замеченными неврологами, если не достигают значительной выраженности. Тем не менее, без этого аспекта картина клинических проявлений болезни не может считаться полной [3].
Основные виды психических нарушений у детей с эпилепсией, столь же многочисленные и многообразные, как пароксизмальные проявления болезни, можно схематически отнести к одной из 4 категорий: 1) астенические состояния (невротические реакции астенического типа); 2) нарушения психического развития (с различной выраженностью интеллектуального дефицита); 3) девиантные формы поведения; 4) аффективные расстройства [1, 8, 9, 11].
Наиболее типичными изменениями личности при определенной длительности течения эпилепсии считается полярность аффекта (сочетание аффективной вязкости склонности «застревать» на тех или иных, особенно отрицательно окрашенных, аффективных переживаниях, с одной стороны, и аффективной взрывчатости, импульсивности с большой силой аффективного разряда — с другой); эгоцентризм с концентрацией всех интересов на собственных потребностях и желаниях; аккуратность, доходящая до педантизма; гиперболизированное стремление к порядку, ипохондричность, cочетание грубости и агреcсивности по отношению к одним с угодливостью и подобострастием к другим лицам (например, к старшим, от которых больной зависит) [1, 3, 8, 11].
В дополнение к этому, для детей и подростков с эпилепсией свойственны патологические изменения сферы инстинктов и влечений (повышенный инстинкт самохранения, повышение влечений, с чем связаны жестокость, агрессивность, иногда — повышенная сексуальность), а также темперамента (замедление темпа психических процессов, преобладание хмурого и угрюмого настроения) [1, 10].
Менее специфичными в картине стойких изменений личности при эпилепсии являются нарушения интеллектуально-мнестических функций (замедленность и тугоподвижность мышления — брадифрения, его персеверативность, склонность к детализации, снижение памяти по эгоцентрическому типу и т. д.); описываемые изменения становятся более заметными у детей, достигших возраста начала школьного обучения [1, 8, 11].
Вообще же среди психических отклонений, свойственных эпилепсии, фигурируют следующие нарушения: рецепторные расстройства, или сенсопатии (сенестопатии, гиперестезия, гипестезия); расстройства восприятия (галлюцинации: зрительные, экстракампинные, слуховые, вкусовые, обонятельные, тактильные, висцеральные, гипнагогические и комплексные; псевдогаллюцинации); психосенсорные расстройства (дереализация, деперсонализация, изменение скорости течения событий во времени); аффективные нарушения (гипер- и гипотимия, эйфория, экстатические состояния, дисфория, паратимии, апатия; неадекватность, диссоциация и полюсность аффекта; страхи, аффективное исключительное состояние, аффективная неустойчивость и др.); расстройства памяти или дисмнезия (амнезия антероградная, ретроградная, антероретроградная и фиксационная; парамнезии); нарушение внимания (расстройства концентрации внимания, «застреваемость» внимания, суженное внимание); расстройства интеллекта (от темповой задержки психомоторного развития до деменции); нарушения моторики (гипер- и гипокинезия); речевые расстройства (афазия моторная, сенсорная или амнестическая; дизартрия, олигофазия, брадифазия, речевой автоматизм и т. д.); так называемые «расстройства побуждений» (мотивации): гипер- и гипобулия; расстройства влечения (анорексия, булимия, навязчивости); нарушения сна или диссомнии (гиперсомния, гипосомния); психопатоподобные расстройства (характерологическое нарушение эмоционально-волевых функций и поведения); различные формы дезориентации (во времени, окружении и собственной личности) [1, 3, 8, 9–12].
Практически все описанные выше нарушения могут приводить или сопровождаться теми или иными нарушениям сознания. Поэтому в «эпилептопсихиатрии» А. И. Болдаревым (2000) в первую очередь рассматриваются синдромы изменения сознания: синдром повышения ясности сознания и синдромы снижения ясности сознания (парциальные и генерализованные) [12].
Синдром повышения ясности сознания (или синдром сверхбодрствования). Встречается при эпилепсии довольно часто, хотя остается малоизученным. Содержание синдрома повышения ясности сознания определяют следующим образом: ясность, живость и отчетливость восприятия; быстрая ориентация в окружающем, мгновенность и яркость воспоминаний, легкость разрешения возникшей ситуации, быстрое течение мыслительных процессов, чуткая откликаемость на все происходящее. Считается, что наиболее отчетливо синдром повышения ясности сознания проявляется при гипертимии, а также при гипоманиакальном и экстатическом состояниях [1, 12].
Синдромы снижения ясности сознания парциальные. При эпилепсии являются переходными состояниями между сохранным и глубоко нарушенным сознанием пациента. Они могут возникать в пред-, меж- или постприступном периодах и довольно многообразны (снижение восприимчивости внешних стимулов и раздражителей, нарушения их ассоциативной переработки, заторможенность различной степени выраженности, транзиторное снижение интеллекта, замедленность реакций и психических процессов, снижение коммуникабельности, притупление эмоций, сужение объема внимания, нарушение воспоминаний, а также частичное расстройство ориентации во времени, окружении и собственной личности и т. д.). К «особым состояниям сознания» А. И. Болдарев (2000) относит психосенсорные расстройства и изменения восприятия во времени (включая феномены déjà vu и jamais vu). При эпилепсии сноподобные состояния (dreamy states) являются нередким вариантом парциального расстройства сознания (по типу jamais vu или déjà vu); их продолжительность варьирует от нескольких секунд или минут до нескольких часов/суток. Сноподобные состояния характерны для височной эпилепсии. Эпилептические трансы — немотивированное и необоснованное перемещение пациента из одного места в другое, происходящее на фоне частичного расстройства сознания и внешне упорядоченного поведения, а также последующей неполной амнезии. Трансы различной продолжительности (от нескольких часов до нескольких недель) могут провоцироваться эмоциональным стрессом и/или острой соматической патологией (ОРЗ и т. д.) [1, 12].
Синдромы снижения ясности сознания генерализованные сравнительно многочисленны. К ним принято относить следующие психопатологические феномены: оглушенность (затруднение и замедление образования/воспроизведения ассоциаций); делирий (расстройство сознания, насыщенное зрительными и/или слуховыми галлюцинациями с последующей неполной амнезией); онейроид (сновидное состояние, при котором грезоподобные события происходят в субъективном нереальном пространстве, но воспринимаются как реальные); просоночные состояния (изменение сознания и неполная ориентация в происходящем или отсутствие ориентации и бодрствования после пробуждения); сомнамбулизм (хождение в ночное время в состоянии неполного сна); простые психомоторыне припадки (кратковременные — по несколько секунд, одиночные автоматические действия с выключением сознания) и сложные психомоторные припадки (более продолжительные — до 1 минуты и более, приступы автоматизма с выключением сознания, напоминающие кратковременные сумеречные состояния); сумеречные состояния сознания (полная дезориентация пациента, аффективная напряженность, галлюцинации, бредовая интерпретация происходящего, возбуждение, неадекватное и немотивированное поведение); аментивные состояния (глубокое нарушение ориентации в окружающем и собственной личности в сочетании с неспособностью к образованию и воспроизведению ассоциаций; после выхода больного из аментивного состояния отмечается полная амнезия); сопорозное состояние (глубокое нарушение сознания, из которого пациента можно вывести на непродолжительное время резким раздражением — кратковременное частичное прояснение сознания; при выходе из сопорозного состояния отмечается антероградная амнезия); кома (глубокое бессознательное состояние с отсутствием реакции на внешние раздражители — зрачковый и корнеальный рефлексы не определяются; после выхода из коматозного состояния имеет место антероградная амнезия); ундулирующее расстройство сознания (перемежающиеся колебания сознания — от ясного до полного его выключения) [1, 12].
Другие психические расстройства при эпилепсии, встречающиеся в детском возрасте, представлены следующими нарушениями: синдром дереализации (нарушения пространственного восприятия во время приступов); синдромы нарушения восприятия во времени (déjà vu, jamais vu, déjà entendu (уже слышанное)); синдром сочетания психосенсорных расстройств с частичным изменением сознания, нарушением восприятия во времени и экстатическим состоянием (психосенсорные расстройства — деперсонализационные и дереализационные, включая нарушения схемы тела, экстатическое состояние, нереальность времени и т. д.); синдром психосенсорных расстройств и онейроидного состояния (комплексный синдром грубой дереализации, деперсонализации и онейроида); синдром неопределенности субъективных переживаний (невозможность конкретизировать собственные субъективные ощущения и переживания, иногда со слуховыми или зрительными галлюцинациями); синдром диссоциации между объективными и субъективными переживаниями (отрицание пациентом наличия многоформных или абортивных эпилептических приступов, отмечающихся как ночью, так и в дневное время); комплексные синдромы (сложные приступы с сочетанием различных ощущений, висцеровегетативных проявлений, аффективных нарушений и других симптомов); бредовые синдромы (паранойяльный, параноидный или парафренный); кататоническое субступорозное состояние (неполная обездвиженность при затяжных и хронических эпилептических психозах, нередко сочетающаяся с частичным или полным мутизмом, мышечным гипертонусом и явлениями негативизма); кататонические синдромы (кататоническое возбуждение — импульсивность, манерность, неестественность, двигательное возбуждение, или ступор — мутизм, каталепсия, эхолалия, эхопраксия, стереотипия, гримасничанье, импульсивные акты); синдром Кандинского–Клерамбо или синдром психического автоматизма (псевдогаллюцинации, психические автоматизмы, бред преследования и воздействия, чувство овладения и открытости; возможны 3 варианта психического автоматизма: ассоциативный, кинестетический и сенестопатический); синдром психической расторможенности или гиперкинетический синдром (общая расторможенность с быстро сменяющимися движениями, неусидчивость, невозможность концентрации внимания, повышенная отвлекаемость, непоследовательность в действиях, нарушения логического построения, непослушание) [1, 8, 12].
Когнитивные нарушения при эпилепсии
Нарушения когнитивных функций встречаются при парциальных и генерализованных формах эпилепсии. Характер когнитивного «эпилептического» дефицита может быть приобретенным, флюктуирующим, прогрессирующим, хроническим и деградирующим (приводящим к развитию деменции) [1, 12].
- Deonna и E. Roulet-Perez (2005) выделяют 5 групп основных факторов, потенциально объясняющих когнитивные (и поведенческие) проблемы у детей при эпилепсии: 1) патология головного мозга (врожденная или приобретенная); 2) эпилептогенное повреждение; 3) эпилепсия как основа электрофизиологической дисфункции; 4) влияние лекарственных препаратов; 5) воздействие психологических факторов [13].
Структура интеллекта у больных с эпилепсией характеризуется нарушением восприятия, снижением концентрации внимания, объема кратковременной и оперативной памяти, моторной активности, зрительно-моторной координации, конструктивного и эвристического мышления, скорости формирования навыков и т. д., что обусловливает у пациентов трудности в социальной интеграции и образовании, снижая качество жизни. Негативное влияние на когнитивные функции раннего дебюта эпилепсии, рефрактерности к проводимой терапии, токсического уровня антиэпилептических препаратов в крови продемонстрировано многими исследователями [1, 12, 14, 15].
Cимптоматические эпилепсии вследствие органического повреждения ЦНС также являются серьезным фактором риска по нарушениям когнитивных функций. Нарушения высших психических функций при эпилепсии зависят от локализации очага эпилептической активности и/или структурного повреждения мозга. При левостороннем повреждении у детей с лобной эпилепсией отмечается дефицит решительности, вербальной долговременной памяти, затруднения в зрительно-пространственном анализе. Частые приступы у них влияют на уровень внимания и способность к торможению импульсивных ответов; пациенты с дебютом эпилепсии в возрасте до 6 лет не способны к построению поведенческой стратегии [1, 12, 14].
При генерализованной эпилепсии эпилептиформные изменения на ЭЭГ вызывают транзиторные нарушения когнитивных функций (удлинение времени реакции и др.) [1, 12].
Грубые нарушения когнитивных функций свойственны эпилептическим энцефалопатиям раннего детского возраста (ранняя миоклонус-энцефалопатия, синдромы Отахары, Веста, Леннокса–Гасто и др.). Комплексные парциальные приступы, правополушарная локализация эпилептогенного фокуса снижают поддержание (устойчивость) внимания, а феномен ЭЭГ-паттерна продолженной пик-волновой активности в фазу медленно-волнового сна влияет на избирательность и распределение внимания [1, 3].
Прогрессирующая нейрональная ишемия является одной из предпосылок эпилептогенеза, как следствие хронической сосудистой недостаточности. Изменения церебральной перфузии могут служить функциональным субстратом нарушений когнитивных/психофизиологических функций [1, 12].
Большинство антиэпилептических препаратов может вызывать психотропные эффекты (тревога и нарушения настроения, косвенно нарушающие когнитивные функции) [16]. Негативными эффектами этих препаратов являются снижение внимания, ухудшение памяти и скорости психических процессов и т. д. T. A. Ketter и соавт. (1999) высказали гипотезу о различных профилях антиэпилептического и психотропного действия (седативного, стимулирующего или смешанного) препаратов, используемых в лечении эпилепсии [17].
Литература
- Эпилепсия в нейропедиатрии (коллективная монография) / Под ред. Студеникина В. М. М.: Династия. 2011, 440 с.
- Epileptic syndromes in infancy, childhood and adolescence (Roger J., Bureau M., Dravet Ch., Genton P. et al., eds.). 4 th ed. (with video). Montrouge (France). John Libbey Eurotext. 2005. 604 p.
- Encyclopedia of basic epilepsy research / Three-volume set (Schwartzkroin P., ed.). 1–3. Philadelphia. Elsevier/Academic Press. 2009. 2496 p.
- Chapman K., Rho J. M. Pediatric epilepsy case studies. From infancy and childhood through infancy. CRC Press/Taylor&Francis Group. Boca Raton–London. 2009. 294 p.
- Berger H. An unusual manifestation of epilepsy: fever // Postgrad. 1966. Vol. 40. P. 479–481.
- Lin K. L., Wang H. S. Reverse Shapiro’s syndrome: an unusual cause of fever of unknown origin // Brain Dev. 2005. 27. P. 455–457.
- Dundar N. O., Boz A., Duman O., Aydin F. et al. Spontaneous periodic hypothermia and hyperhidrosis // Pediatr. 2008. Vol. 39. P. 438–440.
- Ковалев В. В. Эпилепсия. Глава XIX. В кн.: Психиатрия детского возраста: Руководство для врачей. Изд-е 2-е., перераб. и дополн. М.: Медицина. 1995. С. 482–520.
- Dunn D. W. Neuropsychiatric aspects of epilepsy in children // Epilepsy Behav. Vol. 4. P. 98–100.
- Austin J. K., Dunn D. W. Progressive behavioral changes in children with epilepsy // Prog. Brain Res. 2002. Vol. 135. P. 419–427.
- Болдарев А. И. Психические особенности больных эпилепсией. М.: Медицина. 2000. 384 с.
- Балканская С. В. Когнитивные аспекты эпилепсии в детском возрасте. В кн: Проблемы детской неврологии / Под ред. Г. Я. Хулупа, Г. Г. Шанько. Минск: Харвест. 2006. С. 62–70.
- Deonna T., Roulet-Perez E. Cognitive and behavioural disorders of epileptic origin in children. Mac Keith Press. 2005. 447 p.
- Sanchez-Carpintero R., Neville B. G. Attentional ability in children with epilepsy // Epilepsia. Vol. 44. S. 1340–1349.
- Tromp S. C., Weber J. W., Aldenkamp A. P., Arends J. et al. Relative influence of epileptic seizures and of epilepsy syndrome on cognitive function // J. Child Neurol. Vol. 18. P. 407–412.
- Aldenkamp A. P. Effects of antiepileptic drugs on cognition // Epilepsia. Vol. 42. Suppl. 1. S. 46–49.
- Ketter T. A., Post R. M., Theodore W. H. Positive and negative psychiatric effects of antiepileptic drugs in patients with seizure disorders // Neurology. Vol. 53. P. 53–67.
Источник: Журнал «Лечащий врач» №8, 2014
Существует немало форм эпилепсии, встречающихся исключительно в детском или подростковом возрасте. Именно зависимость от возраста многих разновидностей эпилепсии является главным отличительным признаком эпилептологии детского возраста [1–4].
Эпилепсии и судорожные синдромы периода новорожденности
Хотя продолжительность неонатального периода невелика, целый ряд эпилептических синдромов свойственен именно для новорожденных детей [3–5].
Доброкачественные семейные приступы (судороги) новорожденных
Доброкачественная неонатальная эпилепсия (с аутосомно-доминатным типом наследования) трех типов, проявляющаяся в первые 7 дней жизни (начиная с трех суток). В семейном анамнезе обязательно фигурируют указания на наличие в прошлом судорог у членов семьи пациента (в неонатальном периоде). Связь припадков с уточненными врожденными нарушениями метаболизма не установлена. Доброкачественные семейные неонатальные приступы манифестируют в виде фокальных и мультифокальных или генерализованных тонико-клонических (судорожных) припадков. Указанные припадки характеризуются малой продолжительность (1–2 мин) и значительной частотой (20–30 эпизодов за сутки). Впоследствии, по прошествии от 1 до 3 недель, приступы самопроизвольно спонтанно купируются.
Доброкачественные несемейные судороги (приступы) новорожденных («припадки пятого дня»)
Эта эпилепсия с дебютом в раннем неонатальном периоде имеет также другое название (доброкачественные идиопатические неонатальные судороги). Болезнь впервые описана в конце 1970-х гг. Судорожные приступы развиваются у доношенных новорожденных детей, не имевших до этого признаков патологии со стороны ЦНС. Дебют приступов происходит к концу 1-й недели жизни (в 80–90% случаев — между 4-м и 6-м днями), а их пик приходится на 5-й день жизни (отсюда и название). Описываемые приступы обычно имеют вид мультифокальных клонических судорог, которым нередко сопутствуют апноэ. В большинстве случаев доброкачественные идиопатические неонатальные судороги длятся не более 24 ч (они всегда прекращаются по прошествии 15 дней после дебюта). В 80% случаев за время судорожного периода у новорожденных отмечается развитие эпилептического статуса [3–5].
Ранняя инфантильная эпилептическая энцефалопатия с паттерном «угнетение/вспышка» на ЭЭГ (синдром Отахары)
Ранняя инфантильная эпилептическая энцефалопатия — редкая болезнь, относящаяся к злокачественным формам эпилепсии детского возраста. Дебютирует обычно в периоде новорожденности (или в возрасте 1–3 мес). Болезнь характеризуется тоническими приступами, частота которых значительно варьирует (10–300 эпизодов за сутки). У детей отмечается быстрое формирование неврологического дефицита и задержка психического развития. Специфический паттерн «вспышка/угнетение» при электроэнцефалографии (ЭЭГ) представлен у детей c синдромом Отахары как в состоянии сна, так и при бодрствовании. При магнитно-резонансной томографии (МРТ) головного мозга у пациентов отмечаются грубые аномалии развития ЦНС. Среди детей с ранней инфантильной эпилептической энцефалопатией с паттерном «вспышка/угнетение» на ЭЭГ летальность к возрасту 1 года достигает 40–50%. В 4–6-месячном возрасте синдром Отахары может трансформироваться в синдром Веста [3–6].
Ранняя миоклоническая (эпилептическая) энцефалопатия
Описана J. Aicardi и F. Goutières (1978); дебютирует преимущественно в периоде новорожденности (иногда до 3-месячного возраста). В генезе болезни предполагается роль генетических факторов и некоторых «врожденных ошибок метаболизма» (пропионовая ацидурия, метилмалоновая ацидемия, болезнь мочи с запахом кленового сиропа и др.). Клинически проявляется частыми миоклоническими припадками. Последние обычно не ассоциированы с ЭЭГ-изменениями во время приступа, но в ряде случаев одновременно с миоклониями регистрируются эпилептиформные разряды «угнетение/вспышка». Миоклонии чаще бывают фрагментарными (легкие подергивания дистальных отделов конечностей, век или углов рта); одновременно могут отмечаться фокальные (парциальные) приступы, массивные миоклонии и тонические спазмы (изолированные или серийные — возникают к 3–4 месяцам). Появление у ребенка тонических спазмов заставляет предположить наличие атипичного синдрома Веста, но вскоре основные проявления болезни возобновляются и сохраняются на протяжении длительного времени. Фокальные припадки (сложные парциальные — с заведением глаз или автономными симптомами: апноэ, гиперемия лица; клонические судороги в разных участках тела и др.) становятся основным типом приступов при ранней миоклонической эпилептической энцефалопатии. При интериктальном ЭЭГ-исследовании у детей регистрируется паттерн «угнетение/вспышка», состоящий из разрядов продолжительностью 1–5 сек, чередующийся с почти изоэлектрическими периодами (длительностью 3–10 сек). Описываемый ЭЭГ-паттерн становится более отчетливым во время сна (особенно в фазе глубокого сна). Изначальный паттерн «угнетение/вспышка» по достижении возраста 3–5 мес сменяется атипичной гипсаритмией или мультифокальными пароксизмами, но в большинстве случаев это лишь транзиторный феномен. Болезнь сопровождается высокой летальностью или прогрессивным распадом психомоторных функций (вплоть до вегетативного статуса), хотя по мере увеличения возраста частота и выраженность фокальных приступов и миоклоний постепенно уменьшаются [3–5, 7].
Витамин В6-зависимая эпилепсия
Cравнительно редкое наследственное заболевание, характеризующееся фармакорезистентными судорогами. Относится к группе метаболически обусловленных эпилепсий. Развивается у новорожденных, матери которых длительно получали пиридоксин во время беременности, а также при специфическом наследственном дефекте метаболизма (с повышенной потребностью в витамине В6). Известны случаи дебюта пиридоксинзависимых судорог у детей старше 1 мес и даже на втором году жизни. Между приступами судорог дети остаются беспокойными, реагируют мышечными подергиваниями на внешние раздражения. Болезнь не поддается обычному противосудорожному лечению, но назначение витамина В6 в высоких дозах (25 мг/кг/сут) быстро приводит к нормализации состояния [3–5].
Злокачественные мигрирующие парциальные судороги (приступы) младенческого возраста
Чрезвычайно редко встречающийся эпилептический синдром, описанный G. Coppola и соавт. (1995). К настоящему времени сообщается всего о примерно 50 случаях болезни, зарегистрированных в различных странах мира. Злокачественные мигрирующие парциальные судороги в 50% случаев наблюдаются в первые дни жизни; остальные 50% приходятся на возраст 1–3 мес. При дебюте приступы носят фокальный клонический характер, а по прошествии нескольких недель они становятся мультифокальными, причем исключительно частыми и фармакорезистентными к терапии антиэпилептическими препаратами. При ЭЭГ-исследовании у детей выявляется выраженная многоочаговая эпилептическая активность; метаболических нарушений не обнаруживается, а МРТ-признаки патологических изменений отсутствуют. Паталогоанатомическое исследование позволило выявить в гиппокампе признаки нейрональной потери [1, 3, 5, 8].
Эпилепсии у детей первого года жизни (1–12 мес)
По достижении 1-месячного возраста число разновидностей эпилептических синдромов, специфичных для первого года жизни ребенка, практически не уступает таковому, свойственному периоду новорожденности.
Инфантильные спазмы (синдром Веста)
Этот вариант катастрофической эпилепсии (генерализованной) бывает симптоматическим (подавляющее большинство случаев) или криптогенным (10–20%). Он манифестирует у детей на первом году жизни (чаще между 3-м и 8-м месяцами). В классическом варианте синдром Веста характеризуется в момент приступа комбинацией сгибательных и разгибательных движений, то есть выраженными миоклоническими (салаамовыми) спазмами, иногда серийными короткими сгибательными движениями головы («кивки»). Инфантильные спазмы могут развиться как вследствие наличия различной неврологической патологии, так и без каких-либо очевидных предшествующих нарушений функций ЦНС. При инфантильных спазмах психомоторное развитие замедляется, в дальнейшем высока вероятность выраженного отставания в развитии. В 80% случаев при синдроме Веста обнаруживаются микроцефалия, признаки детского церебрального паралича, атонически-атактические нарушения и др. Отличительным электрофизиологическим признаком синдрома Веста является гипсаритмия (по данным ЭЭГ), которая имеет вид диффузных высоковольтажных пиков и медленных волн, располагающихся на дезорганизованном (медленном) фоне. Прогноз синдрома Веста определяется эффективностью проводимой терапии, но в целом малоблагоприятен [3–8].
Тяжелая миоклонус-эпилепсия младенческого возраста (синдром Драве)
Болезнь, описанная C. Dravet (1978, 1992), дебютирует на первом году жизни (между 2-м и 9-м мес), что нередко происходит вслед за развитием фебрильного эпизода, вскоре после вакцинации или перенесения инфекции. Синдром Драве характеризуется появлением генерализованных или односторонних клонических судорог (обычно на фоне гипертермии или лихорадки), что происходит на фоне предшествующего нормального психомоторного развития ребенка на протяжении первого года жизни. Постепенно (по прошествии нескольких недель или месяцев) у ребенка развиваются афебрильные миоклонические и парциальные (фокальные) припадки. Прогрессивное нарастание частоты миоклоний (изолированных или серийных) предшествует появлению у пациентов генерализованных припадков. У детей выявляются умеренные мозжечковые и пирамидные знаки, связанные с дефицитарностью грубой моторики и атаксией походки. Нарушения психомоторного развития впоследствии отмечаются у детей примерно до 4-летнего возраста. Нередко при синдроме Драве у детей развивается эпилептический статус (судорожный или миоклонический). Данные ЭЭГ на протяжении первого года жизни обычно остаются в пределах нормы, хотя у отдельных пациентов встречаются спонтанные фотоиндуцированные пик-волновые разряды. Впоследствии иктальные ЭЭГ-исследования при синдроме Драве характеризуются наличием миоклонических или клонических припадков (генерализованная пик-волновая или полипик-волновая активность). Генерализованные разряды усиливаются в состоянии релаксации; одновременно отмечаются фокальные и мультифокальные пики и острые волны. Традиционные и новые антиэпилептические препараты обычно не предотвращают рецидива приступов при синдроме Драве. Прогноз по интеллектуальному развитию при синдроме Драве всегда неблагоприятен [3–5, 8].
Идиопатические доброкачественные парциальные эпилепсии младенчества
Обычно дебютируют у детей в возрасте 3–20 месяцев (чаще между 5-м и 8-м мес). Впервые описаны K. Watanabe и соавт. (1987), вследствие чего изначально получили обобщающее название «синдром Ватанабе». Характеризуются проявлениями в виде сложных парциальных (фокальных) приступов и благоприятным прогнозом (элиминация эпилептических припадков в течение 3 мес после дебюта). В среднем число приступов составляет около 7; у части пациентов отмечаются исключительно сложные парциальные припадки, у других — только вторично-генерализованные, а примерно в половине случаев встречается их сочетание. Во время приступа для пациентов характерны снижение реакции на предъявляемые стимулы, остановка двигательной активности, умеренные судорожные подергивания, латеральное заведение глаз и цианоз. Основными клиническими признаками этой группы эпилепсий являются высокая встречаемость кластерных приступов, короткая продолжительность припадков, а также изначально нормальные показатели интериктального ЭЭГ-исследования (впоследствии у части детей могут обнаруживаться пароксизмальные разряды) [2, 3, 5, 6, 8].
Сходные с идиопатическими доброкачественными парциальными эпилепсиями младенчества, но исключительно семейные пароксизмальные состояния с дебютом на первом году жизни носят название «доброкачественные инфантильные семейные судороги». В 1997 г. были описаны сходные с ними случаи семейных эпилепсий с последующим формированием хореоатетоза — семейные судороги с хореатетозом [3–5, 8, 9].
Эпилепсии у детей раннего возраста (1–3 года)
Для детей раннего возраста (от 12 до 36 месяцев), в первую очередь, характерны cиндром Доозе, синдром Леннокса–Гасто, доброкачественная миоклонуc-эпилепсия младенческого возраста, синдром гемиконвульсий-гемиплегии, идиопатическая парциальная эпилепсия младенчества, абсансная эпилепсия раннего детства, электрический эпилептический статус медленно-волнового сна, ранний и поздний детский нейрональный липофусциноз (типы I и II).
Миоклоническая астатическая эпилепсия раннего детского возраста (cиндром Доозе)
Представляет собой эпилепсию c миоклонически-астатическими приступами (различной продолжительности). Приступы дебютируют в возрасте 1–5 лет. Чаще болезнь поражает мальчиков. Астатические и миоклонические приступы могут сочетаться, причем миоклонии возникают как до, во время, так и после астатического припадка. Приступы наступают внезапно и практически всегда сопровождаются падениями. Миоклонии отмечаются в виде различной выраженности симметричных подергиваний в руках и мышцах плеч пояса, что сочетается с наклоном головы («кивки»). Признаки утраты сознания у детей в момент приступа отсутствуют. До начала заболевания психомоторное развитие детей обычно соответствует норме. У части детей болезнь осложняется риском развития деменции (предположительно за счет развития эпилептического статуса абсансов). При ЭЭГ регистрируются генерализованные билатерально-синхронные комплексы пик-волн (3 и более за 1 сек, 2–4 Гц). Прогноз при миоклонически-астатической эпилепсии раннего детского возраста малоблагоприятен [3–6, 8].
Синдром Леннокса–Гасто, или миокинетическая эпилепсия раннего детства с медленными пик-волнами
Группа гетерогенной патологии с эпилептическими приступами (атоническими, тоническими, атипичными абсансами), интеллектуальной дефицитарностью и характерным ЭЭГ-паттерном. Как и при синдроме Веста, при синдроме Леннокса–Гасто выделяют симптоматический и криптогенный варианты болезни. Ранние формы дебютируют примерно с 2-летнего возраста. До 30% случаев представляют собой результат трансформации из синдрома Веста. Клинически синдром Леннокса–Гасто характеризуется миоклонически-астатическими припадками, салаамовыми судорогами (молниеносными кивательными), атипичными абсансами, тоническими приступами (чаще во сне). Могут встречаться генерализованные тонико-клонические, миоклонические и фокальные (парциальные) приступы. Для детей типична серийность припадков с изменениями сознания (ступор) и постепенным переходом в эпилептический статус. Помимо эпилептических приступов, в неврологическом статусе могут отмечаться церебральные парезы/параличи, а также атонически-астатические нарушения (до 40% пациентов). У детей происходит снижение интеллекта (различной степени), наблюдаются выраженные нарушения когнитивных функций. По ЭЭГ-данным типичны изменения фоновой активности в виде медленных пик-волн < 3 Гц, ночные серии пиков (гипсаритмия с наличием «острых» феноменов). Часто присутствуют мультифокальные изменения. Методы нейровизуализации позволяют выявить фокальные и диффузные структурные нарушения. До 75–80% случаев болезни резистентны к проводимой терапии. Прогноз вариабелен, но в целом считается малоблагоприятным [3–6, 8, 9].
Доброкачественная миоклонуc-эпилепсия младенческого возраста
Дебютирует у детей до 3 лет (чаще в 1–2 года), хотя иногда может манифестировать с возраста 4–11 месяцев. До появления болезни обычно не отмечается предшествующих признаков нарушений психомоторного развития. Относится к генерализованным/идиопатическим или симптоматическим эпилепсиям. Отличается от синдрома Драве меньшей выраженностью и более благоприятным прогнозом. Для доброкачественной миоклонус-эпилепсии младенчества характерно появление кратковременных миоклонических приступов, вовлекающих голову и верхние конечности. Другие типы приступов при этой форме эпилепсии не возникают, а частота и интенсивность имеющихся пароксизмов вариабельна. Возможна умеренная задержка интеллектуального развития. Диагноз устанавливается на основании клинических особенностей приступов, а также по данным ЭЭГ. При иктальном ЭЭГ-исследовании определяется генерализованная эпилептическая активность с нерегулярными пиками, пик-волнами, острыми волнами (чаще асимметричными, чем билатерально-синхронными); интериктально данные ЭЭГ либо не изменены, либо нарушены умеренно (острые волны, пики, комплексы пик-волна, острая/медленная волна — с преобладанием в ранних стадиях сна) [3–6, 8].
Синдром гемиконвульсий-гемиплегии (HHS)
Болезнь чаще дебютирует в раннем возрасте (1–3 года), хотя возможно начало болезни в период с 1-го по 4-й год жизни (начиная примерно с 5 мес). Первыми проявлениями являются внезапно развившиеся длительные гемиконвульсии в виде клонических судорог (статус), которые иногда (не во всех случаях) бывают связаны с гипертермией (фебрильные судороги). После судорожного эпизода у детей отмечается гемиплегия. По прошествии 12–36 мес у пациентов развивается фокальная (парциальная) эпилепсия. При ЭЭГ у детей отмечается пик- и полипик-медленноволновая активность с частотой 10–12 Гц (преимущественно в затылочных отведениях). В ряде случаев синдром гемиконвульсий-гемиплегии рассматривается в качестве нетипичного следствия продолжительных фебрильных судорог в младенческом и/или раннем детском возрасте [3–5].
Доброкачественная парциальная эпилепсия младенческого и раннего детского возраста с вертексными пиками и волнами во время сна
Выделена итальянскими эпилептологами G. Capovilla и F. Beccaria (2000) в отличие от доброкачественной парциальной эпилепсии младенческого возраста, описанной К. Watanabe и соавт. (1987). Она дебютирует в 13–30 месяцев, ЭЭГ-изменения (характерные пики и волны в период медленного сна с локализацией в вертексных отделах мозга) наблюдаются только во время сна (ЭЭГ при пробуждении всегда не показывает эпилептиформных изменений), клиническая картина приступов также имеет некоторые отличия. Прогноз при доброкачественной парциальной эпилепсии младенческого и раннего детского возраста с вертексными пиками и волнами во время сна благоприятен [3, 5, 8, 9].
Абсансная эпилепсия раннего детства
Этот эпилептический синдром, название которого было предложено H. Doose и соавт. (1965), включает гетерогенную группу пациентов. Абсансная эпилепсия раннего детства клинически характеризуется генерализованными клонико-тоническими судорогами и/или миоклонически-астатическими приступами, наличием нерегулярных пик-волновых разрядов при ЭЭГ (2–3 Гц), нередко — неблагоприятным прогнозом. Абсансная эпилепсия раннего детства иногда дебютирует в более позднем возрасте — до 5 лет [3–6, 8].
Миоклонии век с абсансами (синдром Дживонса)
Возрастзависимый эпилептический синдром, являющийся формой фотосенситивной эпилепсии, описал P. M. Jeavons (1977). Может дебютировать раньше, чем детская абсансная эпилепсия (в 2–5 лет), хотя наиболее часто встречается в возрасте 6–8 лет. Несколько чаще встречается у девочек. У большинства пациентов в семейном анамнезе имеются указания на наличие идиопатической генерализованной эпилепсии. J. Guiwer и соавт. (2003) подчеркивают, что синдром Дживонса является скорее миоклоническим, чем абсансным синдромом. Хотя синдром Дживонса относится к идиопатическим генерализованным эпилепсиям, не исключено, что в это понятие входит целая группа фотосенситивных состояний. Главным признаком синдрома Дживонса являются миоклонии век, а основным триггер-фактором болезни служит закрытие глаз при наличии освещения. Абсансы при синдроме Дживонса наблюдаются не всегда; они имеют короткую продолжительность (по 3–6 сек), появляются после закрытия глаз и сопровождаются четким ритмическим паттерном миоклоний век, а также ретропульсией глазных яблок в ассоциации с тоническим компонентом вовлеченных в патологический процесс мышц. Абсансы при синдроме Дживонса не появляются без миоклоний век. В большинстве случаев у детей наблюдаются генерализованные тонико-клонические припадки, но частота этих приступов сравнительно невысока. При ЭЭГ у пациентов регистрируются электрофизиологические пароксизмы, ассоциированные с закрытием глаз и фотосенситивностью. Нарушения сознания при синдроме Дживонса не столь выражены, как при детской абсансной эпилепсии или ювенильной абсансной эпилепсии. Интеллектуальное развитие при миоклониях век с абсансами практически не страдает, хотя в отдельных случаях сообщается о легком или умеренном интеллектуальном дефиците. Диагноз легко подтверждается при помощи видео-ЭЭГ, поскольку выявляет сопряженность генерализованной пароксизмальной активности с закрытием глаз. В целом прогноз при синдроме Дживонса благоприятен, хотя эта форма эпилепсии чаще оказывается пожизненной. Известны случаи формирования фармакорезистентности болезни [3–6, 9].
Ранний детский (инфантильный) нейрональный цероидный липофусциноз тип I (классический)
Представитель группы прогрессирующих миоклонус-эпилепсий. Связан с дефицитом фермента α-нейраминидазы (сиалидазы), известен также под названием «синдром вишневой косточки с миоклонусом» или «болезни Сантавуори–Халтиа». Может проявляться с возраста от 6 до 24 мес. Основными клиническими проявлениями болезни являются миоклонус и генерализованные тонико-клонические судорожные припадки, к которым впоследствии присоединяется атаксия с расстройством походки. Для болезни характерно наличие на глазном дне симптома «вишневой косточки» и прогрессирующее снижение зрения (вплоть до слепоты). В ряде случаев интеллектуальное развитие детей не страдает, но у части пациентов отмечается формирование деменции. При помощи методов нейровизуализации может быть выявлена диффузная атрофия гемисфер коры головного мозга и мозжечка. Терапия заключается в применении антимиоклонических препаратов, ноотропов, левокарнитина, витаминов (Е, А, группы В) [3, 5, 8, 9].
Поздний детский (инфантильный) нейрональный цероидный липофусциноз тип II
Как и ранний детский нейрональный липофусциноз, поздняя форма болезни относится к прогрессирующим миоклонус-эпилепсиям. Этот вид патологии ранее был известен также под названием «болезнь Янского–Бильшовского», или «поздняя детская амавротическая идиотия». Поздний детский нейрональный цероидный липофусциноз тип II обычно проявляется позднее, чем ранний (тип I), c конца 1-го года жизни до 2–3-летнего возраста, хотя описаны и врожденные формы заболевания. Для болезни типичны миоклонические приступы и атаксия. Помимо описанного поражения ЦНС, у детей страдают и другие внутренние органы и опорно-двигательный аппарат (нейросенсорная тугоухость, грыжи — паховые, мошоночные, пупочные; гепатоспленомегалия, патологическая подвижность суставов, постепенно сменяющаяся на ограниченность, и т. д.). При офтальмологическом исследовании выявляется симптом «вишневой косточки», а также помутнение роговицы. Характерны глубокие расстройства интеллекта. Диагноз подтверждается исследованием активности нейраминидазы в свежих фибробластах и лейкоцитах. При ЭЭГ у больных регистрируется быстрая низковольтажная активность, хотя при деменции встречается ее замедление. Генерализованные пик-волновые вспышки отсутствуют или встречаются редко. Принципы лечения, используемые при позднем детском нейрональном липофусцинозе типа II, cсоответствуют таковым при ранней форме болезни типа I [3, 5, 6, 9].

Комментировать