
Продолжение статьи. Обязательные и дополнительные критерии диагностики. Диагностические критерии атопического дерматита у детей, согласно американским специалистам.
Д. Ш. Мачарадзе, доктор медицинских наук, профессор, РУДН, Москва
Атопический дерматит не имеет специфических лабораторных маркеров диагностики, и потому диагностика заболевания основывается в основном на характерных клинических признаках.
В 1980 г. J. Hanifin и G. Rajka опубликовали большие и малые критерии атопического дерматита, которые оказали неоценимое влияние на научную и клиническую медицинскую практику, хотя считается, что они больше пригодны для использования в стационарных условиях, чем для популяционных исследований.
Диагностический алгоритм атопического дерматита, предложенный американскими экспертами в 1999 г. и основанный на критериях Hanifin и Rajka, включает обязательные и дополнительные критерии [1].
Обязательные критерии:
- зуд кожных покровов;
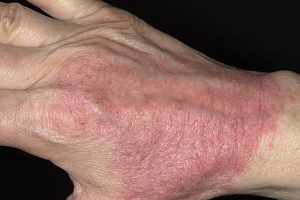
- типичная морфология и локализация кожных высыпаний (у детей);
- экзематозные кожные высыпания, локализующиеся на лице и разгибательных поверхностях конечностей; у взрослых — лихенизация и экскориации на сгибательных поверхностях конечностей);
- хроническое рецидивирующее течение;
- атопия в анамнезе или наследственная предрасположенность к атопии.
Дополнительные критерии:
- ксероз (сухость) кожи;
- ладонный ихтиоз;
- реакция немедленного типа при кожном тестировании с аллергенами;
- локализация кожного процесса на кистях и стопах;
- экзема сосков;
- восприимчивость к инфекционным поражениям кожи, связанная с нарушением клеточного иммунитета;
- начало заболевания в раннем детском возрасте;
- эритродермия;
- рецидивирующий конъюнктивит;
- складки Денье–Моргана (суборбитальные складки);
- кератоконус (коническое выпячивание роговицы);
- передние субкапсулярные катаракты;
- высокий уровень иммуноглобулина Е (IgE) в сыворотке крови.
Нижеперечисленные симптомы также включают в диагностический алгоритм атопического дерматита [2], поскольку они встречаются у большинства таких больных: хейлит, трещины за ушами, экзема лица, шелушение кожи без экземы, плантарный дерматоз.
Диагноз атопического дерматита считается доказанным, если у больного выявлены три и более обязательных и дополнительных признаков.
Критерии Hanifin и Rajka сохраняют свою силу и вполне подходят для клинической диагностики обеих форм атопического дерматита. Как известно, согласно номенклатуре Европейского респираторного общества, различают две формы заболевания: extrinsic, которая связана с сенсибилизацией к внешним аллергенам и характеризуется повышением уровня IgE; intrinsic, при которой не удается подтвердить наличие атопии, несмотря на типичную клиническую картину поражения кожи.
Американские специалисты предлагают более упрощенные диагностические критерии атопического дерматита у детей [3].
Обязательные: зуд, экзематозные изменения, хроническое или рецидивирующее течение.
Типичная для данного возраста локализация кожного поражения: вовлечение лица, шеи и разгибательных поверхностей конечностей у младенцев и детей; поражение сгибательных поверхностей, особенно у детей более старшего возраста и взрослых; щадящее — паховых областей и подмышечных впадин.
Важные особенности (свидетельствуют в пользу данного диагноза, но встречаются не у всех пациентов):
- начало заболевания в раннем возрасте;
- ксероз;
- атопия (IgE-реактивность).
Как показывает практика, для атопического дерматита характерен клинический полиморфизм. Достаточно сказать, что в литературе описано более 26 отдельных мест локализации кожного процесса на участках тела и 9 типичных признаков атопического дерматита. Выделяют, наряду с сухостью кожи, три основных признака: зуд, эритема, папулы. У детей клинические проявления заболевания, особенно локализация кожных повреждений, зависят от возраста. Так, в младенческом периоде (с рождения до 2 лет) поражаются щеки, лицо, шея, наружная поверхность конечностей. В детском возрастном периоде (от 2 до 10 лет) высыпания обычно локализуются в локтевых и подколенных складках, на спине, задней поверхности шеи, боковых поверхностях туловища. Для подросткового и взрослого периода характерно поражение лица (в основном периорбитально или по типу атопического хейлита), тыльных поверхностей кистей, в области локтевых и коленных сгибов. Зудящие папулы располагаются на фоне лихенизированной и сухой кожи, нередко с выраженным шелушением.
Дифференциальная диагностика
Даже тщательно разработанные обязательные и дополнительные критерии атопического дерматита Hanifin и Rajka не позволяют во всех случаях точно диагностировать болезнь. Практика показывает, что применение данного алгоритма более целесообразно при проведении дифференциальной диагностики атопического дерматита и других кожных заболеваний, к которым российские эксперты относят [4]:
- себорейный дерматит;
- контактный дерматит;
- иммунодефицитные заболевания (синдром Вискотта–Олдрича, синдром Джоба);
- микробную экзему;
- розовый лишай;
- чесотку;
- нарушение обмена триптофана.
В одном из последних руководств по аллергическим заболеваниям американские специалисты предлагают следующий спектр для дифференциальной диагностики атопического дерматита у детей [5]:
- атопический дерматит;
- энтеропатический акродерматит;
- гаммаглобулинемию;
- атаксию-телеангиэктазию;
- болезнь Хартнупа;
- гипер-IgE-синдром;
- фенилкетонурию;
- скабиес;
- себорейный дерматит;
- синдром Вискотта–Олдрича.
Ведущий американский специалист по атопическому дерматиту профессор D. Leung рекомендует исключать у больных подросткового возраста также кожную Т-клеточную лимфому и аллергический контактный дерматит [6]. По мнению D. Leung, в плане дифференциальной диагностики всегда важно помнить о том, что самой главной причиной неудачного исхода в терапии атопического дерматита является низкий уровень комплаентности.
Остановимся более подробно на заболеваниях, с которыми рекомендуют проводить дифференциальную диагностику атопического дерматита американские специалисты и которые плохо освещены в отечественной литературе в качестве нозологий. Однако о них следует помнить при исключении диагноза атопического дерматита у детей.
Энтеропатический акродерматит. Это аутосомно-рецессивное заболевание, чаще встречается у девочек и начинается на первом году жизни. Везикулобуллезные и экзематозные высыпания располагаются симметрично, чаще вокруг рта, на щеках, коленных и локтевых суставах, а также в перианальной области. Со временем эти эритематозные и эрозивные элементы становятся сухими, гиперкератическими и даже напоминают псориаз. Для этого заболевания характерна алопеция, а волосы отличаются своеобразным рыжеватым оттенком. Другими характерными признаками энтеропатического акродерматита являются хроническая диарея и различные проявления со стороны глаз (фотофобия, конъюнктивит, блефарит, дистрофия роговицы), стоматит, паронихии, отставание в росте и интеркуррентные бактериальные и грибковые инфекции. Заболевание связывают с дефицитом цинка и нарушением его абсорбции в кишечнике. Для диагностики энтеропатического акродерматита исследуют концентрации цинка в плазме крови и щелочной фосфатазы, цинкозависимого фермента.
Агаммаглобулинемия. Первичное иммунодефицитное состояние, которое проявляется у мальчиков в возрасте 6–12 мес жизни повторными бактериальными пневмониями, а также высокой частотой хронических и рецидивирующих гнойно-воспалительных инфекций ЛОР-органов, кишечника, глаз, кожи, лимфоузлов. Лабораторная диагностика основывается на выявлении (минимум двукратном) сывороточной концентрации IgG < 200 мг%, IgA и IgM < 20 мг% и почти полном отсутствии В-лимфоцитов.
Атаксия-телеангиэктазия является комплексным синдромом комбинированного иммунодефицита и наследуется как аутосомно-рецессивный признак. В клинической картине заболевания отмечаются кожные, печеночные, неврологические и эндокринные расстройства [7]. У больных появляется телеангиэктазия конъюнктивы и кожи (к 3–6 годам), рецидивирующие бактериальные инфекции придаточных пазух носа и легких; различные дефекты клеточного и гуморального звеньев иммунитета, повышается частота возникновения злокачественных новообразований. В типичных случаях атаксия возникает вскоре после того, как ребенок начинает ходить, и к 10–12 годам жизни становится резко выраженной.
Болезнь Хартнупа (нарушение обмена триптофана) — наследственная ферментопатия триптофанового обмена. Первые эритематозные или иногда везикулезные элементы у ребенка появляются уже в первые месяцы жизни на лице, руках и ногах в «виде перчаток и носков». Могут присоединяться гиперпигментация и гиперкератоз, усиливающийся под воздействием солнечного света. Зуд кожи может быть разной степени выраженности. Нарушение всасывания триптофана в кишечнике ведет в школьном возрасте (вследствие нарушения синтеза никотинамида) сначала к развитию отдельных симптомов и лишь позднее к слабоумию и грубой симптоматике органического поражения центральной нервной системы (мозжечковая атаксия, судороги, снижение интеллекта и др.). Параллельно у больных нарастает пеллагроподобная сыпь с фотосенсибилизацией. Для заболевания характерно также наличие врожденных изменений костной системы. Сопутствующими заболеваниями являются реактивный панкреатит, гепатит, синдром мальабсорбции и т. д. В крови отмечается эозинофилия, высокий уровень общего IgE. Болезнь Хартнупа диагностируют с учетом данных, полученных на консультации у генетика, а также в результате специального обследования (хроматография аминокислот мочи и крови, нагрузочные тесты с триптофаном, тест на ксантуреновую кислоту).
Фенилкетонурия (болезнь Феллинга). Внедрение неонатального скрининга на фенилкетонурию (хроматографическое определение фенилаланина в моче) позволяет с высокой степенью достоверности выявить заболевание уже на 5-й день жизни ребенка. Характерные симптомы болезни: слабоумие всех степеней, частые судорожные припадки, светлые волосы, предрасположенность к экземе, «запах конюшни».
Иммунодефицитные заболевания (синдром Вискотта–Олдрича, синдром гипер-IgE). Согласно рекомендациям Всемирной организации здравоохранения (ВОЗ), первичный иммунодефицит подозревают у детей, имеющих:
- частые отиты (не менее 6–8 раз в течение одного года);
- несколько подтвержденных синуситов, протекавших в тяжелой форме (не менее 4–6 раз в течение одного года);
- не менее 2 раз перенесенные и подтвержденные пневмонии;
- повторные глубокие абсцессы кожи или внутренних органов;
- необходимость в длительной терапии антибиотиками для купирования инфекции (до 2 мес и дольше);
- необходимость во внутривенных антибиотиках для купирования инфекции;
- перенесенные не менее 2 раз глубокие инфекции, такие как менингит, остеомиелит, целлюлит, сепсис;
- отставание грудного ребенка в росте и весе;
- персистирующую молочницу или грибковое поражение кожи в возрасте старше 1 года;
- наличие у родственников первичных иммунодефицитов, ранних смертей от тяжелых инфекций или одного из вышеперечисленных симптомов [8].
Заболевания (в том числе аллергические), ассоциированные с первичным иммунодефицитом, диагностируют на основании соответствующих клинико-диагностических критериев и иммунологического обследования больного (определение уровней Ig A, M, G (включая подклассы IgG, IgE), активности комплементарного, фагоцитарного компонентов, клеточного звена иммунной системы и т. д.). Диагноз аллергического заболевания (бронхиальная астма, аллергический ринит, атопический дерматит, пищевая аллергия) ставят на основании характерных клинических симптомов, а также положительных данных кожных проб и/или повышенного уровня специфических IgE-антител в сыворотке крови.
Из более чем 70 описанных первичных иммунодефицитных состояний дерматит или высокий уровень сывороточного IgE являются обязательными компонентами двух заболеваний: синдрома Вискотта–Олдрича и синдрома Джоба (или синдрома гипер-IgE) [7, 8].
Синдром Вискотта–Олдрича — Х-сцепленное заболевание, входит в группу комбинированной недостаточности клеточного и гуморального иммунитета, наследуется как аутосомно-рецессивная патология. Синдром Вискотта–Олдрича достаточно редкое заболевание: частота его встречаемости составляет 4:1 000 000 родившихся живыми мальчиков [7].
Заболевание проявляется у детей в возрасте первых месяцев жизни до 3 лет и характеризуется триадой: дерматитом, похожим на атопический, рецидивирующими инфекциями желудочно-кишечного и респираторного трактов и тромбоцитопенией.
Молекулярно-генетическим исследованием обнаружено отсутствие белка WASP (Wiscott-Aldrich syndrom protein), кодируемого геном 11-й пары хромосом. Тромбоцитопению объясняют различными причинами: повышенным разрушением клеток; аномальным метаболизмом; неэффективным тромбоцитопоэзом [7]. Помимо классической, выделена и более легкая форма синдрома, названная Х-сцепленной тромбоцитопенией, которая протекает без выраженных признаков иммунодефицита.
В клинической картине синдрома Вискотта–Олдрича обращают внимание на признаки тромбоцитопении, которые выявляются с первых месяцев жизни ребенка (удлинение времени кровотечения из ранки, кишечные и носовые кровотечения, гематурия, петехии, экхимозы) и которые могут привести к тяжелой анемии [9]. У таких детей инфекционный процесс чаще всего вызван пневмококками и проявляется в виде отитов, пневмонии, сепсиса, менингита. Если дети выживают, в дальнейшем преобладают оппортунистические инфекции, возбудителями которых являются Pneumocystis carini, вирусы герпеса. Нередко у детей старшего возраста обнаруживаются признаки аутоиммунных заболеваний (васкулиты, тромбоцитопения, нейтропения, гломерулонефрит, неспецифический язвенный колит, артрит, узловая эритема) [7–9].
При лабораторном исследовании в крови выявляют снижение числа Т-лимфоцитов, уменьшение их пролиферативной реакции в ответ на антиген; эозинофилию; тромбоцитопению (менее 10% от нормы), уменьшение размеров и функциональную неполноценность тромбоцитов. Уровень сывороточного IgG не изменен (или несколько ниже нормы), IgM умеренно снижен, а концентрации IgA и IgE повышены [10].
Помимо классической триады, у пациентов, страдающих синдромом Вискотта–Олдрича, встречаются рецидивирующий неспецифический артрит, бронхиальная астма, воспалительные заболевания кишечника. Общее состояние таких детей тяжелое, основные причины смерти — рецидивирующие вирусные, грибковые, бактериальные инфекции и кровотечения.
В 2001 г. в отечественной литературе была опубликована статья, в которой приводится тщательное описание опыта диагностики синдрома Вискотта–Олдрича и его паллиативной терапии у 8 детей в возрасте 2–14 лет [11].
Синдром Джоба — синдром гипериммуноглобулинемии Е — аутосомно-доминантное заболевание, с которым ученые ассоциируют еще два других генетических синдрома — пентасомию Х и синдром Дубовица [12].
Патофизиология гипер-IgE-синдрома недостаточно ясна. Одно из объяснений — выраженные нарушения в продукции цитокинов, что затрагивает не только иммунную систему, но и влияет на созревание скелета, синтез коллагена, костный мозг, метаболизм костей [7, 13]. Считается, что у таких больных снижается чувствительность В-лимфоцитов к интерлейкину-4 (ИЛ-4) (главный стимулятор продукции IgE) или же при этом синдроме повышается концентрация противоположного цитокина — интерферона g (ИФНg). По последним данным, в патогенез гипер-IgE-синдрома включается ИЛ-12, который способствует повышению продукции ИФНg [13]. У детей синдром гипер-IgE характеризуется клинической картиной атопического дерматита, также сочетающегося с рецидивирующими гнойными инфекциями (подкожные абсцессы, фурункулез, гнойные отиты), пневмониями, кандидозом кожи и слизистых [7].
В современных публикациях, посвященных синдрому Джоба, минимальными критериями заболевания принято считать наличие у ребенка:
- повышенного уровня сывороточного IgE;
- гнойных стафилококковых инфекций легких;
- экзематозных высыпаний [14].
Синдром гипер-IgE у взрослых больных клинически диагностируют по таким признакам, как экзематозная сыпь, рецидивы пневмоний с образованием абсцесса легкого, повышение сывороточного уровня IgE. Обычно у таких больных не находят лимфоаденопатию и гепатоспленомегалию.
- Grimbacher и соавторы у большинства больных с синдромом гипер-IgE описали также некоторые дисморфические признаки лица, сколиоз, абсцессы кожи, задержку развития зубов [15].
Диагноз заболевания ставят на основе повторного (минимум двукратного) выявления сывороточной концентрации общего IgE > 1000 МЕ/мл при наличии в анамнезе дерматита и повторных глубоких гнойных инфекций с «холодным» течением. У таких больных в анамнезе отмечаются повторные «холодные» абсцессы кожи и подкожной клетчатки, лимфоузлов, повторные гнойные отиты с «холодным» течением [7, 10]. Особую опасность представляют тяжелые эпизоды острых пневмоний, в том числе деструктивных (у 50%) с исходом в пневмоцеле (у 50%); абсцессы печени.
Заболевание иногда действительно трудно отличить от атопического дерматита, особенно у детей — при обеих патологиях у больных могут встречаться такие симптомы, как экзематозный зуд, высыпания; атопия, стафилококковая инфекция, высокий уровень IgE, эозинофилия.
Однако сыпь при атопическом дерматите редко появляется у детей младше 4 мес; она обычно локализуется на лице, щеках, лбу, наружной поверхности конечностей и ягодиц, тогда как у более старших детей — в локтевых и подколенных складках, на кистях и стопах. Сыпь при гипер-IgE-синдроме наблюдается у детей более раннего возраста, располагается на лице, чаще на разгибательных, чем сгибательных поверхностях тела.
Кроме того, у больных с синдромом гипер-IgE чаще наблюдаются инфекции паренхиматозных органов (в основном в виде стафилококковых легочных инфекций), тогда как пациенты, страдающие тяжелым атопическим дерматитом, могут иметь суперинфекцию Staphylococcus (S.) aureus, осложняющую течение экземы, и регионарный лимфаденит. В крови больных гипер-IgE-синдромом выявляют специфические IgE-антитела к S. aureus, Candida albicans, Aspergillus fumigatus. При обеих патологиях очень часто высеивают S. aureus с кожи больных. У взрослых больных с синдромом гипер-IgE часто приходится проводить дифференциальную диагностику с аллергическим бронхолегочным аспергиллезом.




Комментировать